UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
FORM
(Mark One)
For the fiscal year ended
or
For the transition period from to
Commission file number:
(Exact name of registrant as specified in its charter)
| ||
(State or other jurisdiction of | (I.R.S. Employer | |
(Address of principal executive offices and zip code) | ||
(
(Registrant’s telephone number, including area code)
Securities registered pursuant to Section 12(b) of the Act:
Title of each class | Trading Symbol | Name of each exchange on which registered |
Securities registered pursuant to Section 12(g) of the Act:
None
Indicate by check mark if the registrant is a well-known seasoned issuer, as defined in Rule 405 of the Securities Act. Yes ◻
Indicate by check mark if the registrant is not required to file reports pursuant to Section 13 or Section 15(d) of the Act. Yes ◻
Indicate by check mark whether the registrant (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934, as amended, during the preceding 12 months (or for such shorter period that the registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days.
Indicate by check mark whether the registrant has submitted electronically every Interactive Data File required to be submitted pursuant to Rule 405 of Regulation S-T (§232.405 of this chapter) during the preceding 12 months (or for such shorter period that the registrant was required to submit such files).
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, a smaller reporting company or an emerging growth company. See the definitions of “large accelerated filer,” “accelerated filer,” “smaller reporting company” and “emerging growth company” in Rule 12b-2 of the Exchange Act.
⌧ | Accelerated filer | ◻ | |||
Non-accelerated filer | ◻ | Smaller reporting company | |||
Emerging growth company |
If an emerging growth company, indicate by check mark if the registrant has elected not to use the extended transition period for complying with any new or revised financial accounting standards provided pursuant to Section 13(a) of the Exchange Act. ◻
Indicate by check mark whether the registrant has filed a report on and attestation to its management’s assessment of the effectiveness of its internal control over financial reporting under Section 404(b) of the Sarbanes-Oxley Act (15 U.S.C. 7262(b)) by the registered public accounting firm that prepared or issued its audit report.
Indicate by check mark whether the registrant is a shell company (as defined in Rule 12b-2 of the Exchange Act). Yes
The aggregate market value of the voting and non-voting common stock held by non-affiliates of the registrant as of the last business day of the registrant’s most recently completed second fiscal quarter was $
As of February 24, 2022, the number of outstanding shares of the registrant’s common stock, par value $0.01 per share, was
DOCUMENTS INCORPORATED BY REFERENCE
Specified portions of the registrant’s proxy statement with respect to the 2022 Annual Meeting of Shareholders, which is to be filed pursuant to Regulation 14A within 120 days after the end of the registrant’s fiscal year ended December 31, 2021, are incorporated by reference into Part III of this Form 10-K.
SPECIAL NOTE REGARDING FORWARD-LOOKING STATEMENTS
This Annual Report on Form 10-K contains forward-looking statements within the meaning of Section 27A of the Securities Act of 1933 (the “Securities Act”) and Section 21E of the Securities Exchange Act of 1934 (the “Exchange Act”), which are subject to the “safe harbor” created by those sections for such statements. Forward-looking statements are based on our management’s beliefs and assumptions and on currently available information. All statements other than statements of historical fact are “forward-looking statements.” Terms such as “anticipate,” “believe,” “could,” “estimate,” “expect,” “goal,” “intend,” “likely,” “may,” “plan,” “possible,” “potential,” “predict,” “project,” “should,” “target,” “will,” “would,” and similar expressions and variations thereof are intended to identify forward-looking statements, but these terms are not the exclusive means of identifying such statements. Examples of these statements include, but are not limited to, statements regarding:
| ● | our estimates regarding how long our existing cash, cash equivalents, short-term investments and revenues will fund our anticipated operating expenses, capital expenditures and debt service obligations; |
| ● | our expectations related to future milestone and royalty payments potentially payable to us under the terms of the asset purchase agreement under which we divested our former commercial ophthalmology product OMIDRIA® (phenylephrine and ketorolac intraocular solution); |
| ● | our expectations regarding clinical plans and anticipated or potential paths to regulatory approval of narsoplimab by the U.S. Food and Drug Administration (“FDA”) and the European Medicines Agency (“EMA”) in hematopoietic stem cell transplant-associated thrombotic microangiopathy (“HSCT-TMA”), immunoglobulin A (“IgA”) nephropathy, and atypical hemolytic uremic syndrome (“aHUS”); |
| ● | whether and when a marketing authorization application (“MAA”) may be filed with the EMA for narsoplimab in any indication, and whether the EMA will grant approval for narsoplimab in any indication; |
| ● | our plans for the commercial launch of narsoplimab following any regulatory approval and our estimates and expectations regarding coverage and reimbursement for any approved products; |
| ● | our expectation that we will rely on contract manufacturers to manufacture narsoplimab, if approved, for commercial sale and to manufacture our drug candidates for purposes of clinical supply and in anticipation of potential commercialization; |
| ● | our expectations regarding the clinical, therapeutic and competitive benefits and importance of our drug candidates; |
| ● | our ability to design, initiate and/or successfully complete clinical trials and other studies for our drug candidates and our plans and expectations regarding our ongoing or planned clinical trials, including for our lead MASP-2 inhibitor, narsoplimab, and for our other investigational candidates, including OMS527 and OMS906; |
| ● | the severity and duration of the impact of the COVID-19 pandemic on our business, operations, clinical programs and financial results; |
| ● | our plans and expectations regarding development of narsoplimab for the treatment of critically ill COVID-19 patients, including statements regarding the therapeutic potential of narsoplimab for the treatment of COVID-19, discussions with government agencies regarding narsoplimab for the treatment of COVID-19, expectations for the treatment of additional COVID-19 patients in clinical trials or other settings and our expectations for receiving any regulatory approval or authorization from FDA or other regulatory body for narsoplimab in the treatment of COVID-19 patients; |
i
| ● | with respect to our narsoplimab clinical programs, our expectations regarding: whether enrollment in any ongoing or planned clinical trial will proceed as expected; whether we can capitalize on the financial and regulatory incentives provided by orphan drug designations granted by the FDA, the European Commission (“EC”), or the EMA; and whether we can capitalize on the regulatory incentives provided by fast-track or breakthrough therapy designations granted by FDA; |
| ● | our ability to raise additional capital through the capital markets or through one or more corporate partnerships, equity offerings, debt financings, collaborations, licensing arrangements or asset sales; |
| ● | our expectations about the commercial competition that our drug candidates, if commercialized, face or may face; |
| ● | the expected course and costs of existing claims, legal proceedings and administrative actions, our involvement in potential claims, legal proceedings and administrative actions, and the merits, potential outcomes and effects of both existing and potential claims, legal proceedings and administrative actions, as well as regulatory determinations, on our business, prospects, financial condition and results of operations; |
| ● | the extent of protection that our patents provide and that our pending patent applications will provide, if patents are issued from such applications, for our technologies, programs, and drug candidates; |
| ● | the factors on which we base our estimates for accounting purposes and our expectations regarding the effect of changes in accounting guidance or standards on our operating results; and |
| ● | our expected financial position, performance, revenues, growth, costs and expenses, magnitude of net losses and the availability of resources. |
Our actual results could differ materially from those anticipated in these forward-looking statements for many reasons, including the risks, uncertainties and other factors described in Item 1A of Part I of this Annual Report on Form 10-K under the heading “Risk Factors” and in Item 7 of Part II under the heading “Management’s Discussion and Analysis of Financial Condition and Results of Operations” and in our other filings with the Securities and Exchange Commission (“SEC”). Given these risks, uncertainties and other factors, actual results or anticipated developments may not be realized or, even if substantially realized, they may not have the expected consequences to or effects on our company, business or operations. Accordingly, you should not place undue reliance on these forward-looking statements, which represent our estimates and assumptions only as of the date of the filing of this Annual Report on Form 10-K. You should read this Annual Report on Form 10-K completely and with the understanding that our actual results in subsequent periods may materially differ from current expectations. Except as required by applicable law, including the securities laws of the United States and the rules and regulations of the SEC, we assume no obligation to update or revise any forward-looking statements contained herein, whether as a result of any new information, future events or otherwise.
ii
OMEROS CORPORATION
ANNUAL REPORT ON FORM 10-K FOR THE YEAR ENDED DECEMBER 31, 2021
INDEX
iii
PART I
This Annual Report on Form 10-K contains forward-looking statements reflecting our current expectations that involve risks and uncertainties. Actual results may differ materially from those discussed in these forward-looking statements due to a number of factors, including those set forth in the section entitled “Risk Factors” and elsewhere in this Annual Report. Please refer to the special note regarding forward-looking statements at the beginning of this Annual Report on Form 10-K for further information.
ITEM 1. BUSINESS
Overview
Omeros Corporation (“Omeros,” the “Company” or “we”) is an innovative biopharmaceutical company committed to discovering, developing and commercializing small-molecule and protein therapeutics for large-market and orphan indications targeting immunologic diseases, including complement-mediated diseases and cancers related to dysfunction of the immune system, as well as addictive and compulsive disorders.
Our lead drug candidate narsoplimab is the subject of a biologics license application (“BLA”) currently pending before the U.S. Food and Drug Administration (“FDA”) for the treatment of hematopoietic stem cell transplant-associated thrombotic microangiopathy (“HSCT-TMA”). On October 18, 2021, we announced the receipt of a Complete Response Letter (“CRL”) from FDA regarding the BLA. In the CRL, FDA expressed difficulty in estimating the treatment effect of narsoplimab in HSCT-TMA and asserted that additional information will be needed to support regulatory approval. In February 2022, we had a Type A meeting with FDA to discuss the CRL, including each of the review issues that FDA identified as presenting difficulties interpreting the treatment response in the pivotal trial. We are currently awaiting FDA’s response to our rebuttals to each of those review issues. We continue to believe that our BLA, as submitted, merits approval and that the data meet or exceed the threshold for substantial evidence of effectiveness.
We also have multiple Phase 3 and Phase 2 clinical-stage development programs in progress with narsoplimab, which are focused on: complement-mediated disorders, including immunoglobulin A (“IgA”) nephropathy, atypical hemolytic uremic syndrome (“aHUS”) and COVID-19. We are also initiating a Phase 1b clinical program in paroxysmal nocturnal hemoglobinuria (“PNH”) for our MASP-3 inhibitor OMS906 targeting the alternative pathway of complement and have successfully completed a Phase 1 study in our phosphodiesterase 7 (“PDE7”) program focused on addiction. In addition, we have a diverse group of preclinical programs, including GPR174, a novel target in immuno-oncology that modulates a new cancer immunity axis that we discovered. Small-molecule and antibody inhibitors of GPR174 are part of our proprietary G protein-coupled receptor (“GPCR”) platform through which we control 54 GPCR drug targets and their corresponding compounds.
We previously developed and commercialized OMIDRIA® (phenylephrine and ketorolac intraocular solution) 1%/0.3%, which is approved by FDA for use during cataract surgery or intraocular lens (“IOL”) replacement to maintain pupil size by preventing intraoperative miosis (pupil constriction) and to reduce postoperative ocular pain. We marketed OMIDRIA in the United States (“U.S.”) from the time of its commercial launch in 2015 until December 2021.
On December 23, 2021, we completed the sale of OMIDRIA and certain related assets and liabilities to Rayner Surgical Inc. (“Rayner”) pursuant to an Asset Purchase Agreement, dated December 1, 2021 (the “Asset Purchase Agreement”). We received approximately $126.0 million in cash at the closing and we will receive a royalty of 50% of the net revenue, as defined in the Asset Purchase Agreement, from sales of OMIDRIA in the U.S. between the closing date and the earlier of January 1, 2025 or the payment of the $200.0 million milestone described below. After such date, we will receive a royalty of 30% of the net revenue from sales of OMIDRIA in the U.S. until the expiration or termination of the last issued and unexpired patent with respect to OMIDRIA in the U.S. The U.S. base royalty rate is subject to a reduction down to 10% upon the occurrence of certain events described in the Asset Purchase Agreement, including during any specific period in which OMIDRIA is no longer eligible for separate payment (i.e., outside the packaged payment rate for the surgical procedure) under Medicare Part B. We will also will receive a royalty of 15% of the net revenue from sales of OMIDRIA outside the U.S. on a country-by-country basis between the closing date and the expiration or termination of the last issued and unexpired patent with respect to OMIDRIA in such country. In addition,
1
we will receive a $200.0 million milestone payment if, prior to January 1, 2025, separate payment for OMIDRIA is secured under Medicare Part B for a continuous period of at least four years.
We launched OMIDRIA in the U.S. in the second quarter of 2015 and sold OMIDRIA primarily through wholesalers which, in turn, sold to Ambulatory Surgical Centers (“ASC”) and hospitals. The Centers for Medicare & Medicaid Services (“CMS”), the federal agency responsible for administering the Medicare program, granted transitional pass-through reimbursement status for OMIDRIA from January 1, 2015 through December 31, 2017. In March 2018, Congress extended pass-through reimbursement status for OMIDRIA through September 30, 2020 when used during procedures performed on Medicare Part B fee-for-service patients. Pass-through reimbursement for OMIDRIA under Medicare Part B expired on October 1, 2020. In December 2020, in its calendar year 2021 Outpatient Prospective Payments System (“OPPS”) and ASC Payments System final rule, CMS determined that, under its policy applicable to certain non-opioid pain management surgical drugs, OMIDRIA qualifies for separate payment when used on Medicare Part B patients in the ASC setting. CMS’ policy of separately reimbursing non-opioid pain management surgical drugs was first adopted in 2019 and became applicable to OMIDRIA upon the expiration of the drug’s pass-through reimbursement on October 1, 2020. In November 2021, CMS issued its final OPPS and ASC Payments Systems rule for calendar year 2022 which reconfirmed CMS’ policy regarding non-opioid pain management surgical drugs and states that OMIDRIA will continue to receive separate payment when used on Medicare Part B patients in the ASC setting.
Our Drug Candidates and Development Programs
2
Our clinical drug candidates consist of the following:
Drug Candidate/Program |
| Targeted Disease(s) |
| Development Status |
| Next Expected |
| Worldwide | |||
|---|---|---|---|---|---|---|---|---|---|---|---|
Clinical | |||||||||||
Narsoplimab (OMS721/MASP-2) - | Hematopoietic Stem-Cell Transplant-Associated Thrombotic Microangiopathy (HSCT-TMA) | Pivotal Trial Complete; CRL received; BLA pending before FDA | BLA resubmission | Omeros | |||||||
Narsoplimab (OMS721/MASP-2) - | Immunoglobulin A Nephropathy (IgAN) | Phase 3 | Complete Phase 3 patient enrollment and perform 36-week assessment of proteinuria | Omeros | |||||||
Narsoplimab (OMS721/MASP-2) - | Atypical Hemolytic Uremic Syndrome (aHUS) | Phase 3 | Complete Phase 3 patient enrollment | Omeros | |||||||
Narsoplimab (OMS721/MASP-2) | Severe COVID-19 requiring mechanical ventilation | Phase 2 | Read out data from platform clinical trial |
| Omeros (In-licensed) | ||||||
PDE7 (OMS527) | Addictions and compulsive disorders; movement | Phase 1 | Initiate Phase 2 clinical program pending availability of resources | Omeros | |||||||
MASP-3 (OMS906) - | Paroxysmal Nocturnal Hemoglobinuria (PNH) and other alternative pathway disorders | Phase 1 | Initiate Phase 1b clinical trial in PNH patients with suboptimal response to the C5 inhibitor ravulizumab | Omeros | |||||||
PPARγ (OMS405) - | Opioid and nicotine addiction | Phase 2 | Evaluate data from investigator-sponsored trial in patients with cocaine use disorder | Omeros | |||||||
3
Our pipeline of development programs consists of the following:
Drug Candidate/Program |
| Targeted Disease(s) |
| Development Status |
| Next Expected |
| Worldwide | ||||||||||||||
Preclinical / Platform | ||||||||||||||||||||||
MASP-2 - Small- | aHUS, IgAN, HSCT-TMA and age-related macular degeneration | Preclinical | Identify drug development candidate for clinical trials | Omeros | ||||||||||||||||||
MASP-2 – Second Generation Antibody | Long-acting second generation antibody targeting lectin pathway disorders | Preclinical/Phase 1 | CTA submission | Omeros | ||||||||||||||||||
MASP-3 - Small- | PNH and other alternative pathway disorders | Preclinical | Identify drug development candidate for clinical trials | Omeros | ||||||||||||||||||
GPR174 Inhibitors and Related Therapeutics | Wide range of cancers | Preclinical | Identify drug development candidate for clinical trials | Omeros | ||||||||||||||||||
Chimeric Antigen Receptor (CAR) T-Cell and Adoptive T-Cell Therapies | Wide range of cancers | Preclinical | Scale up and clinical trial initiation | Omeros | ||||||||||||||||||
> 50 other GPCR targets | Immunologic, | Preclinical | Identify drug development candidate for clinical trials | |||||||||||||||||||
MASP Inhibitor Clinical Programs
MASP-2 Program - Narsoplimab (OMS721) - Lectin Pathway Disorders
Overview. Mannan-binding lectin-associated serine protease-2 (“MASP-2”), is a novel pro-inflammatory protein target involved in activation of the complement system, which is an important component of the immune system. The complement system plays a role in the body’s inflammatory response and becomes activated as a result of tissue damage or trauma or microbial pathogen invasion. Inappropriate or uncontrolled activation of the complement system can cause diseases characterized by serious tissue injury. Three main pathways can activate the complement system: classical, lectin, and alternative. MASP-2 is recognized as the effector enzyme of the lectin pathway and is required for the function of this pathway. Importantly, inhibition of MASP-2 has been demonstrated not to interfere with the antibody-dependent classical complement activation pathway, a critical component of the acquired immune response to infection.
Our proprietary, patented lead human monoclonal antibody targeting MASP-2, which we have referred to as OMS721, has been assigned the nonproprietary name narsoplimab. The current development focus for narsoplimab is diseases in which the lectin pathway has been shown to contribute to significant tissue injury and pathology. When not treated, these diseases are typically characterized by significant end-organ damage, such as kidney or central nervous system injury. We have completed our pivotal clinical trial for narsoplimab in HSCT-TMA and Phase 3 clinical programs are in process for narsoplimab in IgA nephropathy and aHUS. Narsoplimab is also being evaluated for
4
treatment of COVID-19 in a nationwide adaptive platform trial and has been used under compassionate use to treat COVID-19 patients in Italy and in the U.S.
Thrombotic Microangiopathies
HSCT-TMA. In October 2020, we reported final clinical data from our pivotal trial of narsoplimab in HSCT-TMA, a frequently lethal complication of HSCT. In November 2020, we completed the rolling submission of our BLA for narsoplimab for the treatment of HSCT-TMA and FDA accepted the BLA for filing in January 2021 under its Priority Review program. In October 2021, we received a CRL from FDA regarding the BLA. In the CRL, FDA expressed difficulty in estimating the treatment effect of narsoplimab in HSCT-TMA and asserted that additional information will be needed to support regulatory approval. In January 2022 we submitted a response to the CRL comprising a comprehensive briefing package addressing the points raised by FDA in the CRL and a request for a Type A meeting with FDA to discuss the CRL. In February 2022, we had a Type A meeting with FDA to discuss the CRL, including each of the review issues that FDA identified as presenting difficulties interpreting the treatment response in the pivotal trial. We are currently awaiting FDA’s response to our rebuttals to each of those review issues. We continue to believe that our BLA, as submitted, merits approval and that the data meet or exceed the threshold for substantial evidence of effectiveness.
The final clinical data from our pivotal trial of narsoplimab in HSCT-TMA in October 2020 was obtained from a single-arm, open-label trial The company worked closely with FDA on design of the single-arm trial to support approval and the definition of response as the primary endpoint.
The primary efficacy endpoint in the trial was the proportion of patients who achieved designated “responder” status based on improvement in HSCT-TMA laboratory markers and clinical status. The primary laboratory markers evaluated were platelet count and lactate dehydrogenase (“LDH”) levels, while improvement in clinical status was evaluated based on organ function and transfusions. Each patient was required to show improvement in both laboratory markers and clinical status to be considered a responder. All others were considered non-responders.
Among patients who received at least one dose of narsoplimab, the complete response rate was 61% (95% confidence interval [CI] 40.6 to 78.5; p<0.0001), while the complete response rate among patients who received the protocol-specified narsoplimab treatment of at least four weeks of dosing was 74% (95% CI 51.6 to 89.8; p<0.0001). The response rates and their respective lower levels of the 95% confidence intervals are a multiple of the pre-specified efficacy threshold of 15%.
Secondary endpoints in the trial were survival rates and change from baseline in HSCT-TMA laboratory markers. Among all treated patients, 68% survived for at least 100 days following HSCT-TMA diagnosis, while 83% of patients who received treatment for at least four weeks and 94% of the responders achieved this endpoint. Median overall survival was 274 days among all patients and 361 days among patients who received the protocol-specified treatment of at least four weeks. Median survival could not be estimated for responders because more than half of the responders were alive at last follow-up. Results also included statistically significant improvements in platelet count, LDH and haptoglobin. The treated population had multiple high-risk features that portend a poor outcome, including the persistence of HSCT-TMA despite modification of immunosuppression (which was a criterion for entry into the trial), graft-versus-host disease, significant infections, non-infectious pulmonary complications and neurological findings. The most common adverse events observed in the trial were nausea, vomiting, diarrhea, hypokalemia, neutropenia and fever, which are all common in stem-cell transplant patients. Six deaths occurred during the trial. These were due to sepsis, progression of the underlying disease, and graft-versus-host disease with TMA. All of these are common causes of death in this patient population.
In Europe, the EMA has confirmed narsoplimab’s eligibility for the EMA’s centralized review of a single MAA that, if approved, authorizes the product to be marketed in all EU member states and EEA countries. We are targeting to complete our MAA submission in 2022.
In the U.S., FDA has granted narsoplimab (1) breakthrough therapy designation in patients who have persistent TMA despite modification of immunosuppressive therapy, (2) orphan drug designation for the prevention (inhibition) of
5
complement-mediated TMAs, and (3) orphan drug designation for the treatment of HSCT-TMA. The European Commission (“EC”) also granted narsoplimab designation as an orphan medicinal product for treatment in hematopoietic stem cell transplantation.
aHUS. We have a Phase 3 clinical program in patients with aHUS for which patient recruitment is ongoing. The trial includes multiple sites in the U.S., Asia and Europe; however, enrollment has been slowed due, in large part, to prioritizing the use of resources within our complement programs to narsoplimab in HSCT-TMA and IgA nephropathy, and to OMS906 in PNH. FDA has granted narsoplimab orphan drug designation for the prevention (inhibition) of complement-mediated TMAs and fast-track designation for the treatment of patients with aHUS.
Renal Disease
Phase 3 Program - IgA Nephropathy. Patient enrollment is ongoing in our Phase 3 clinical trial evaluating narsoplimab in IgA nephropathy, which is referred to as ARTEMIS-IGAN. The single Phase 3 trial design is a randomized, double-blind, placebo-controlled multicenter trial in patients at least 18 years of age with biopsy-confirmed IgA nephropathy and 24-hour urine protein excretion greater than 1 g/day at baseline on optimized renin-angiotensin system blockade. This trial includes a run-in period. Initially, patients are expected to receive an IV dose of study drug each week for 12 weeks; additional weekly dosing can be administered to achieve optimal response. The primary endpoint, which could suffice for full or accelerated approval depending on the effect size, is reduction in proteinuria at 36 weeks after the start of dosing. The trial is designed to allow intra-trial adjustment in sample size. For the purposes of safety and efficacy assessments, the initial sample size for the proteinuria endpoint is estimated at 140 patients in each of the treatment and placebo groups. This will include a subset of patients with high levels of proteinuria (i.e., equal to or greater than 2 g/day) at baseline, and a substantial improvement at 36 weeks in this subset of patients alone could potentially form the basis for approval. We believe that the trial design will allow assessment for either full or accelerated approval at 36 weeks based on proteinuria results either (1) across the general population of study patients or (2) in the high-proteinuria subset of patients. In the event of full approval, estimated glomerular filtration rate (“eGFR”) becomes a safety endpoint only. In the event that the primary endpoint at 36 weeks results in accelerated approval from FDA, change in eGFR is expected to be assessed at approximately two years after the start of dosing. These eGFR data, if satisfactory, would then likely form the basis for full approval. In response to investigators’ concerns about extended withholding of narsoplimab treatment from any high-proteinuria patient initially randomized to the placebo-treated group, FDA will allow patients in that sub-population open-label treatment with narsoplimab after at least 1 year of blinded treatment.
In the U.S., narsoplimab has received breakthrough therapy and orphan drug designations from FDA for the treatment of IgA nephropathy. In Europe, narsoplimab has received orphan drug designation from the EMA in patients with IgA nephropathy.
COVID-19.
In March 2020, in response to a request from physicians at the Papa Giovanni XXIII Hospital in Bergamo, Italy, we initiated a compassionate use program for narsoplimab to treat patients with severe COVID-19 requiring mechanical ventilation.
The initial cohort treated under this compassionate use program included a total of six patients with severe COVID-19 treated with narsoplimab under compassionate use, all with acute respiratory distress syndrome (“ARDS”) and requiring continuous positive airway pressure (“CPAP”) or intubation. At baseline, circulating endothelial cell (“CEC”) counts and serum levels of interleukin-6 (“IL-6”), interleukin-8 (“IL-8”), C-reactive protein (“CRP”), LDH, D-dimer and aspartate aminotransferase (“AST”) were markedly elevated.
Narsoplimab treatment was associated with rapid and sustained reduction across all of these markers of endothelial damage and inflammation. In addition, massive bilateral pulmonary thromboses, seen in two of the patients, resolved while on narsoplimab. All six narsoplimab-treated patients recovered, survived and were discharged. Narsoplimab was well tolerated and no adverse drug reactions were reported. Two control groups with similar baseline characteristics were used for retrospective comparison, both showing substantial mortality rates of 32% and 53%. A manuscript
6
detailing the results of the initial cohort of Bergamo patients treated with narsoplimab was published in the peer-reviewed journal Immunobiology. (Rambaldi A, Gritti G, Micò MC, et al. Endothelial injury and thrombotic microangiopathy in COVID-19: Treatment with the lectin-pathway inhibitor narsoplimab. Immunobiology. 2020;225(6):152001.)
All six patients were evaluated five to six months after cessation of narsoplimab treatment. None of them showed any clinical or laboratory evidence of long-term effects of COVID-19 or post-acute sequelae of SARS-CoV-2 infection (“PASC”), such as cognitive impairment or cardiac, pulmonary or other organ disorder, commonly seen following resolution of initial COVID-19 symptoms.
Endothelial damage and resultant thromboses are significant to the pathophysiology of COVID-19, and we believe these data illustrate the importance of inhibiting the lectin pathway to treat critically ill COVID-19 patients. Endothelial damage activates the lectin pathway of complement. We believe the results observed following narsoplimab treatment in critically ill COVID-19 patients at Papa Giovanni were consistent with those seen in HSCT-TMA and underscore the pathophysiologic similarities between these two disorders. Narsoplimab has been shown to inhibit lectin pathway activation and to block the MASP-2-mediated conversion of prothrombin to thrombin, microvascular injury-associated thrombus formation and the activation of factor XII as well as the MASP-2-mediated activation of kallikrein. We believe that the anticoagulant effects of narsoplimab may provide therapeutic benefits in both HSCT-TMA and COVID-19.
Following treatment of the initial six patients under the compassionate use program in Italy, we have continued compassionate-use treatment with 13 more patients in Italy and four patients in the U.S. All of these patients prior to receiving narsoplimab were severely ill, intubated (16) or on CPAP (one), had multiple comorbidities, and had failed other therapies, including anti-virals, targeted anti-inflammatory therapeutics, convalescent plasma and steroids. Following treatment with narsoplimab, the laboratory improvements and clinical outcomes of these patients are similar to those seen in the initial cohort of Bergamo patients.
Two manuscripts from Omeros’ laboratories at the University of Cambridge are expected to be published soon detailing several of our discoveries related to the pathophysiology of COVID-19. The first, submitted for peer-reviewed publication, covers the discovery of a profile of complement markers of broad complement dysfunction seen in all patients examined during the acute phase of severe COVID-19. This dysfunction appears to be driven by hyperactivation of the lectin pathway. Narsoplimab restores complement function in these severe COVID-19 patients while, in patients not treated with narsoplimab, the broad complement dysfunction persists throughout the hospitalization or until death.
The second manuscript, under final review at another peer-reviewed journal, demonstrates that the complement dysfunction in severe COVID-19 patients reported in the first manuscript results in impairment of the adaptive immune response necessary to fight infection, leading to an increased risk of life-threatening secondary infection. Here again treatment with narsoplimab normalizes the adaptive immune response, which should restore the body’s ability to prevent or fight secondary infection and reduce COVID-19 mortality.
Narsoplimab is also the only complement inhibitor included in the I-SPY COVID-19 adaptive platform trial sponsored by Quantum Leap Healthcare Collaborative, which is evaluating drugs and investigational products for the treatment of critically ill COVID-19 patients. The narsoplimab treatment arm of the I-SPY COVID-19 trial has now concluded. Once all data are available, they will be analyzed and the outcome shared publicly.
Discussions regarding the use of narsoplimab in COVID-19 with leaders across various U.S. government agencies continue to progress.
Licensing Arrangements. We hold worldwide exclusive licenses to rights related to MASP-2, the antibodies targeting MASP-2 and the therapeutic applications for those antibodies from the University of Leicester, from its collaborator, the Medical Research Council at Oxford University (“MRC”), and from Helion Biotech ApS (“Helion”). For a more detailed description of these licenses, see “License and Development Agreements” below.
7
MASP-3 Program - OMS906 - Alternative Pathway Disorders
Overview. As part of our MASP program, we have identified mannan-binding lectin-associated serine protease 3 (“MASP-3”), which has been shown to be the key activator of the complement system’s alternative pathway (“APC”), and we believe that we are the first to make this and related discoveries associated with the APC. The complement system is part of the immune system’s innate response, and the APC is considered the amplification loop within the complement system. MASP-3 is responsible for the conversion of pro-factor D to factor D; converted factor D is necessary for the activation of the APC. Based on our alternative pathway-related discoveries, we have expanded our intellectual property position to protect our inventions stemming from these discoveries beyond MASP-2 associated inhibition of the lectin pathway to include inhibition of the alternative pathway. Our current primary focus in this program is developing MASP-3 inhibitors for the treatment of disorders related to the APC. We believe that MASP-3 inhibitors have the potential to treat patients suffering from a wide range of diseases and conditions, including: PNH; multiple sclerosis; neuromyelitis optica; age-related macular degeneration; Alzheimer’s disease; systemic lupus erythematosus; diabetic retinopathy; chronic obstructive pulmonary disease; antineutrophil cytoplasmic antibody-associated vasculitis; anti-phospholipid syndrome; atherosclerosis; myasthenia gravis and others. Our OMS906 monoclonal antibody program has generated positive data in a well-established animal model associated with PNH as well as strong pharmacodynamic activity in non-human primates. The program has also generated positive data in a well-established animal model of arthritis.
In September 2020 we began enrollment and dosing in a placebo-controlled, double-blind, single-ascending-dose and multiple-ascending-dose Phase 1 clinical trial to evaluate the safety, tolerability, pharmacodynamics and pharmacokinetics of OMS906. We have dosed subjects across all dosing cohorts in the single-ascending dose study and reported preliminary data from the Phase 1 trial in June 2021. OMS906 has been well tolerated at all doses tested and preliminary human pharmacokinetic and pharmacodynamic data are consistent with once-monthly subcutaneous dosing and every-other-month or less frequent IV dosing. Recent data show high level suppression of alternative pathway activity. We have determined to forego the multiple-ascending dose portion of our Phase 1 trial in healthy subjects and plan to move directly into a Phase 1b clinical trial in patients with PNH who have an unsatisfactory response to the C5 inhibitor ravulizumab. A successful meeting was held between Omeros and the Medicines and Healthcare products Regulatory Agency (MHRA) to discuss the design and conduct of the Phase 1b trial. Enrollment is expected to begin this summer. We expect that this will accelerate our overall clinical development program for OMS906 in PNH.
Licensing Arrangements. We jointly own and hold worldwide exclusive license rights related to therapeutic applications for inhibiting MASP-3 from the University of Leicester. For a more detailed description of these licenses, see “License and Development Agreements” below.
MASP Inhibitor Preclinical Programs
Other MASP Inhibitor Preclinical Programs
We have generated positive preclinical data from MASP-2 inhibition in in vivo models of AMD, myocardial infarction, diabetic neuropathy, stroke, ischemia-reperfusion injury, and other diseases and disorders.
We are also developing a longer-acting second generation antibody targeting MASP-2, OMS1029 which is expected to enter the clinic this summer. All first-in-human-enabling toxicology studies have been completed, and no findings of concern were identified. Based on pharmacokinetic/pharmacodynamic data to date, dosing in humans is expected to be once-monthly to once-quarterly by subcutaneous or intravenous administration.
Development efforts are also directed to a small-molecule inhibitor of MASP-2 designed for oral administration, as well as small-molecule inhibitors of MASP-3 and bispecific small- and large-molecule inhibitors of MASP-2/-3.
8
Other Clinical Programs
PDE7 Program - OMS527
Overview. Our PDE7 program is based on our discoveries of previously unknown links between PDE7 and any addiction or compulsive disorder, and between PDE7 and any movement disorders, such as Parkinson’s disease. PDE7 appears to modulate the dopaminergic system, which plays a significant role in regulating both addiction and movement. We believe that PDE7 inhibitors could be effective therapeutics for the treatment of addictions and compulsions as well as for movement disorders. Data generated in preclinical studies support the use of PDE7 inhibitors in both of these therapeutic areas.
In September 2019, we reported positive results from our completed Phase 1 clinical trial designed to assess the safety, tolerability and pharmacokinetics of the compound in healthy subjects. In the double blind, randomized Phase 1 study, the study drug, referred to as OMS182399, met the primary endpoints of safety and tolerability and showed a favorable and dose-proportional pharmacokinetic profile supporting once-daily dosing. There was no apparent food effect on plasma exposure to OMS182399. Continued clinical development in our PDE7 program is subject to allocation of financial and other resources, which are currently prioritized for other programs.
Exclusive License Agreement with Daiichi Sankyo Co., Ltd. We hold an exclusive license to certain PDE7 inhibitors claimed in patents and pending patent applications owned by Daiichi Sankyo Co., Ltd. (“Daiichi Sankyo”), as successor-in-interest to Asubio Pharma Co., Ltd., or, for use in the treatment of movement, addiction and compulsive disorders as well as other specified indications. For a more detailed description of our agreement with Daiichi Sankyo, see “License and Development Agreements” below.
PPARγ Program - OMS405
Overview. In our peroxisome proliferator-activated receptor gamma (“PPARγ”) program, we have engaged in development of proprietary compositions that include PPARγ agonists for the treatment and prevention of addiction to substances of abuse, which may include opioids, nicotine and alcohol. We believe that Omeros is the first to demonstrate a link between PPARγ and addiction disorders. Data from clinical studies and from animal models of addiction suggest that PPARγ agonists could be efficacious in the treatment of a wide range of addictions.
Clinical trials. Our collaborators at The New York State Psychiatric Institute have completed two Phase 2 clinical trials related to our PPARγ program. These studies evaluated a PPARγ agonist, alone or in combination with other agents, for treatment of addiction to heroin and to nicotine. The published results of the heroin study demonstrated that, although not altering the reinforcing or positive subjective effects of heroin, the PPARγ agonist significantly reduced heroin craving and overall anxiety. The National Institute on Drug Abuse (“NIDA”) provided substantially all of the funding for these clinical trials and solely oversaw the conduct of these trials. We have the right or expect to be able to reference the data obtained from these studies for subsequent submissions to FDA and continue to retain all other rights in connection with the PPARγ program.
We have also reported positive results (i.e., decreased cravings and protection of brain white matter) from a Phase 2 clinical trial conducted by an independent investigator evaluating the effects of a PPARγ agonist in patients with cocaine use disorder. An investigator-sponsored study evaluating the effects of a PPARγ agonist on the prevention of relapse following treatment of cocaine use disorder is ongoing. The study is funded by NIDA.
Patent Assignment Agreement with Roberto Ciccocioppo, Ph.D. We acquired the patent applications and related intellectual property rights for our PPARγ program in February 2009 from Roberto Ciccocioppo, Ph.D., of the Università di Camerino, Italy, pursuant to a patent assignment agreement. For a more detailed description of our agreement with Dr. Ciccocioppo, see “License and Development Agreements” below.
9
Preclinical Programs and Platforms
GPCR Platform
Overview. GPCRs, which are cell surface membrane proteins involved in mediating both sensory and nonsensory functions, comprise one of the largest families of proteins in the genomes of multicellular organisms. Sensory GPCRs are involved in the perception of light, odors, taste and sexual attractants. Non-sensory GPCRs are involved in metabolism, behavior, reproduction, development, hormonal homeostasis and regulation of the central nervous system. The vast majority of GPCR drug targets are non-sensory. Although GPCRs form a super-family of receptors, individual GPCRs display a high degree of specificity and affinity for the functionally active molecules, or ligands, that bind to a given receptor. Ligands can either activate the receptor (agonists) or inhibit it (antagonists and inverse agonists). When activated by its ligand, the GPCR interacts with intracellular G proteins, resulting in a cascade of signaling events inside the cell that ultimately leads to the particular function linked to the receptor. Without a known ligand, there is no template from which medicinal chemistry efforts can be readily initiated, nor a means to identify the GPCR’s signaling pathway and, therefore, drugs are very difficult to develop against orphan GPCRs. “Unlocking” these orphan GPCRs by identifying one or more of their respective ligands could lead to the development of drugs that act at these new targets.
To our knowledge, Omeros’ technology is the first commercially viable technology capable of identifying ligands of orphan GPCRs in high throughput. We have developed a proprietary cellular redistribution assay (“CRA”), which we use in a high-throughput manner to identify synthetic ligands, including antagonists, agonists and inverse agonists, that bind to and affect the function of orphan GPCRs. We have screened Class A orphan GPCRs against our small-molecule chemical libraries using the CRA and have identified and confirmed compounds that interact with 54 of the 81 Class A orphan GPCRs linked to a wide range of indications including cancer as well as metabolic, cardiovascular, immunologic, inflammatory and central nervous system disorders.
One of our priorities in this program is GPR174, which is involved in the modulation of the immune system. The GPR174 program is part of our immuno-oncology platform. In ex vivo human studies, our small-molecule inhibitors targeting GPR174 upregulate the production of cytokines, block multiple checkpoints and tumor promoters, and suppress regulatory T-cells. Based on our data, we believe that GPR174 controls a major, previously unrecognized pathway in cancer and modulation of the receptor could provide a seminal advance in immuno-oncologic treatments for a wide range of tumors. Our studies in mouse models of melanoma and colon carcinoma found that GPR174-deficiency resulted in significantly reduced tumor growth and improved survival of the animals versus normal mice. Our discoveries suggest a new approach to cancer immunotherapy that targets inhibition of GPR174 and can be combined with and significantly improve the tumor-killing effects of other oncologic agents, including radiation, adenosine pathway inhibitors and checkpoint inhibitors. These discoveries include (1) identification of cancer-immunity pathways controlled by GPR174, (2) the identification of phosphatidylserine as a natural ligand for GPR174, (3) a collection of novel small-molecule inhibitors of GPR174 and (4) a synergistic enhancement of “tumor-fighting” cytokine production by T cells following the combined inhibition of both GPR174 and the adenosine pathway, another key metabolic pathway that regulates tumor immunity. We are developing both small-molecule and antibody inhibitors of GPR174 with the objective of moving compounds into human trials.
In addition to GPR174 inhibitors, we also are developing other cancer therapeutics as well as novel platforms for generating more effective CAR-T and adoptive T-cell therapies.
In addition to Class A orphan GPCRs, we have screened orphan and non-orphan Class B receptors. Class B GPCRs have large extracellular domains and their natural ligands are generally large peptides, making the development of orally active, small-molecule drugs against these receptors, such as glucagon and parathyroid hormone, a persistent challenge. Our CRA technology finds functionally active small molecules for GPCRs, which we believe could lead to the development of oral medications for many of the Class B GPCRs. While our focus to date has remained on Class A orphan GPCRs, we have identified and confirmed sets of compounds that interact selectively with, and modulate signaling of, a small subset of Class B GPCRs, namely glucagon-like peptide-1 receptor and parathyroid hormone 1 receptor.
10
GPCR Platform Funding Agreements with Vulcan Inc. and the Life Sciences Discovery Fund. In October 2010, we entered into funding agreements for our GPCR program with Vulcan Inc. and its affiliate, which we refer to collectively as Vulcan, and with the Life Sciences Discovery Fund Authority (“LSDF”), a granting agency of the State of Washington. For a more detailed description of these agreements, see “License and Development Agreements” below.
Sales and Marketing
We have retained all worldwide marketing and distribution rights to our drug candidates and our development programs. As such, we will be able to market any drug candidate that is approved in the future independently, through arrangements with third parties, or via some combination of these approaches.
We maintained internal marketing and sales capabilities with respect to OMIDRIA until the completion of the divestiture of that product on December 23, 2021. As part of the divestiture, substantially all of our OMIDRIA sales and marketing team members accepted employment with Rayner and were separated from their employment at Omeros, effective as of December 31, 2021.
Manufacturing, Supply and Commercial Operations
We currently do not own or operate manufacturing facilities. We utilized contract manufacturers to produce, store and distribute OMIDRIA and currently rely on third parties to produce sufficient quantities of our drug candidates for use in pre-clinical and clinical studies and for the manufacture of narsoplimab for commercial use following regulatory approval.
OMIDRIA. We assigned or otherwise transitioned to Rayner our agreements with the third parties that produced, stored and distributed OMIDRIA. We required manufacturers that produced active pharmaceutical ingredients (“APIs”) and finished drug products to operate in accordance with current Good Manufacturing Practices (“cGMPs”) and all other applicable laws and regulations.
In the U.S., we sold OMIDRIA through a limited number of wholesalers that distributed the product to ASCs and hospitals. Title transferred upon delivery of OMIDRIA to the wholesaler. We used a single third-party logistics provider to handle warehousing and final packaging of our commercial supply of OMIDRIA in the U.S. and to ship OMIDRIA to our wholesalers. Our third-party logistics provider also performs certain support services on our behalf. Virtually all of our revenues for the last three fiscal years were generated from OMIDRIA product sales in the U.S. Our four major distributors--AmerisourceBergen Corporation, Cardinal Health, Inc., McKesson Corporation and FFF Enterprises, Inc.--together with entities under their common control each accounted for 10% or more, and nearly 100% in aggregate, of our total revenue in 2021. For additional information regarding our major customers, see Part II, Item 8, “Note 2—Significant Accounting Policies” to our Consolidated Financial Statements in this Annual Report on Form 10-K.
Drug Candidates. We have laboratories in-house for analytical method development, bioanalytical testing, formulation, stability testing and small-scale compounding of laboratory supplies of drug candidates. We utilize contract manufacturers to produce sufficient quantities of drug candidates for use in preclinical and clinical studies and to store and distribute our drug candidates. We require manufacturers that produce APIs and finished drug products for clinical use to operate in accordance with cGMPs and all other applicable laws and regulations. We anticipate that we will rely on contract manufacturers to develop and manufacture our drug candidates for commercial sale. We maintain agreements with potential and existing manufacturers that include confidentiality and intellectual property provisions to protect our proprietary rights related to our drug candidates.
In July 2019, we entered into a master services agreement with Lonza Biologics Tuas Pte. Ltd. (“Lonza”) for the commercial production of narsoplimab and for certain regulatory support and related services to be provided by Lonza from time to time. Under the agreement Lonza will manufacture narsoplimab pursuant to purchase orders issued in accordance with forecasts that we provide. We will purchase narsoplimab that meets agreed specifications in batches, with the price per batch varying according to the total number of batches ordered for serial production in a single manufacturing campaign. We are obligated to purchase a minimum number of batches annually beginning on a specified
11
anniversary of the first commercial sale of narsoplimab in either the U.S. or EU. We may be obligated to pay certain fees to Lonza upon cancellation of purchase orders.
The initial term of the agreement expires five years after the first commercial sale of narsoplimab in either the U.S. or EU and is subject to automatic renewal for an additional four-year term unless we provide notice of non-renewal at least three years prior to the end of the initial term. In addition, either party may terminate the agreement, subject to applicable notice and cure periods under certain circumstances. Other than our agreement for commercial supply of narsoplimab, we have not yet entered into a commercial supply agreement for any of our drug candidates.
License and Development Agreements
MASP Program. Under our exclusive license agreements with the University of Leicester and MRC, we have agreed to pay royalties to each of the University of Leicester and MRC that are a percentage of any proceeds we receive from the licensed MASP-2 technology during the terms of the agreements. Our exclusive license agreement with the University of Leicester, but not our agreement with the MRC, also applies to other MASPs. The continued maintenance of these agreements requires us to undertake development activities. We must pay low single-digit percentage royalties with respect to proceeds that we receive from products incorporating certain intellectual property within the licensed technology that are used, manufactured, directly sold or directly distributed by us, and we must pay royalties, in the range of a low single-digit percentage to a low double-digit percentage, with respect to proceeds we receive from sublicense royalties or fees that we receive from third parties to which we grant sublicenses to certain intellectual property within the licensed technology. We did not make any upfront payments for these exclusive licenses nor are there any milestone payments or reversion rights associated with these license agreements. We retain worldwide exclusive licenses from these institutions to develop and commercialize any intellectual property rights developed in the sponsored research. The term of each license agreement ends when there are no longer any pending patent applications, applications in preparation or unexpired issued patents related to any of the intellectual property rights we are licensing under the agreement. Both of these license agreements may be terminated prior to the end of their terms by us for convenience or by one party if the other party (1) breaches any material obligation under the agreement and does not cure such breach after notice and an opportunity to cure or (2) is declared or adjudged to be insolvent, bankrupt or in receivership and materially limited from performing its obligations under the agreement.
In April 2010, we entered into an exclusive license agreement with Helion, pursuant to which we received a royalty-bearing, worldwide exclusive license to all of Helion’s intellectual property rights related to MASP-2 antibodies, polypeptides and methods in the field of inhibition of mannan-binding lectin-mediated activation of the complement system for the prevention, treatment or diagnosis of any disease or condition. We are obligated to make remaining development and sales milestone payments to Helion of up to approximately $5.4 million upon the achievement of certain events, such as receipt of marketing approval, and reaching specified sales milestones. We are obligated to pay Helion a low single-digit percentage royalty on net sales of a MASP-2 inhibitor product covered by the patents licensed under the agreement. The term of the agreement continues so long as there is a valid, subsisting and enforceable claim in any patents or patent applications covered by the agreement. The agreement may be terminated sooner by either party following a material breach of the agreement by the other party that has not been cured within 90 days.
PPARγ. We acquired the patent applications and related intellectual property rights for our PPARγ program in February 2009 from Roberto Ciccocioppo, Ph.D. of the Università di Camerino, Italy, pursuant to a patent assignment agreement. In February 2011, we amended the agreement to include all intellectual property rights, including patent applications, related to nutraceuticals that increase PPARγ activity. Under the amended agreement, we have agreed to pay Dr. Ciccocioppo a low-single digit percentage royalty on net sales of any products that are covered by any patents that issue from the patent applications that we acquired from him. In addition, if we grant any third parties rights to manufacture, sell or distribute any such products, we must pay to Dr. Ciccocioppo a percentage of any associated fees we receive from such third parties in the range of low single-digits to low double-digits depending on the stage of development at which such rights are granted. We have also agreed to make total milestone payments of up to $3.8 million to Dr. Ciccocioppo upon the occurrence of certain development events, such as patient enrollment in a Phase 1 clinical trial and receipt of marketing approval of a drug candidate covered by any patents that issue from the patent applications that we acquired from him. If we notify Dr. Ciccocioppo that we have abandoned all research and development and commercialization efforts related to the patent applications and intellectual property rights we acquired
12
from him, Dr. Ciccocioppo has the right to repurchase those assets from us at a price equal to a double-digit percentage of our direct and indirect financial investments and expenditures in such assets. If he does not exercise his right to repurchase those assets within a limited period of time by paying the purchase price, we will have no further obligations to sell those assets to Dr. Ciccocioppo. The term of our agreement with Dr. Ciccocioppo ends when there are no longer any valid and enforceable patents related to the intellectual property rights we acquired from him, provided that either party may terminate the agreement earlier in case of an uncured breach by the other party. Under the terms of the agreement, we have agreed to pay a portion of the payments due to Dr. Ciccocioppo to the Università di Camerino without any increase to our payment obligations.
PDE7. Under an agreement with Daiichi Sankyo, we hold an exclusive worldwide license to PDE7 inhibitors claimed in certain patents and pending patent applications owned by Daiichi Sankyo for use in the treatment of (1) movement disorders and other specified indications, (2) addiction and compulsive disorders and (3) all other diseases except those related to dermatologic conditions. Under the agreement, we agreed to make milestone payments to Daiichi Sankyo of up to an aggregate total of $33.5 million upon the achievement of certain events in each of these three fields; however, if only one of the three indications is advanced through the milestones, the total milestone payments would be $23.5 million. The milestone payment events include successful completion of preclinical toxicology studies; dosing of human subjects in Phase 1, 2 and 3 clinical trials; receipt of marketing approval of a PDE7 inhibitor drug candidate; and reaching specified sales milestones. In addition, Daiichi Sankyo is entitled to receive from us a low single-digit percentage royalty of any net sales of a PDE7 inhibitor licensed under the agreement by us and/or our sublicensee(s) provided that, if the sales are made by a sublicensee, then the amount payable by us to Daiichi Sankyo is capped at an amount equal to a low double-digit percentage of all royalty and specified milestone payments received by us from the sublicensee.
The term of the agreement with Daiichi Sankyo continues so long as there is a valid, subsisting and enforceable claim in any patents covered by the agreement. The agreement may be terminated sooner by us, with or without cause, upon 90 days advance written notice or by either party following a material breach of the agreement by the other party that has not been cured within 90 days or immediately if the other party is insolvent or bankrupt. Daiichi Sankyo also has the right to terminate the agreement if we and our sublicensee(s) cease to conduct all research, development and/or commercialization activities for a PDE7 inhibitor covered by the agreement for a period of six consecutive months, in which case all rights held by us under Daiichi Sankyo’s patents will revert to Daiichi Sankyo.
GPCR Platform Funding Agreements with Vulcan Inc. and the Life Sciences Discovery Fund. In October 2010, we entered into funding agreements for our GPCR program with Vulcan and LSDF. We received $20.0 million and $5.0 million, respectively, under the agreements with Vulcan and LSDF. Under these agreements, we have agreed to pay Vulcan and LSDF tiered percentages of the net proceeds, if any, that we derive from the GPCR program. The percentage rates of net proceeds payable to Vulcan and LSDF decrease as the cumulative net proceeds reach specified thresholds, and the blended percentage rate payable to Vulcan and LSDF in the aggregate is in the mid-teens with respect to the first approximately $1.5 billion of cumulative net proceeds that we receive from our GPCR program. If we receive cumulative net proceeds in excess of approximately $1.5 billion, the percentage rate payable to Vulcan and LSDF in the aggregate decreases to one percent. An acquirer of the assets in our GPCR program may be required, and an acquirer of our company would be required, to assume all of our payment and other obligations under our agreements with Vulcan and LSDF.
Under our agreement with Vulcan, we granted Vulcan a security interest in our personal property related to the GPCR program, other than intellectual property, which security interest is junior to any existing or future security interests granted in connection with a financing transaction and which will be released automatically after Vulcan receives $25.0 million under the agreement. We also agreed not to grant any liens on intellectual property related to the GPCR program without Vulcan’s consent, subject to specified exceptions. These restrictions could limit our ability to pursue business opportunities involving the GPCR program or reduce the price that a potential buyer would pay for the GPCR assets. If we default under our agreement with Vulcan, in certain circumstances Vulcan may, subject to the rights of any holders of senior security interests, take control of such pledged assets. If we are liquidated, Vulcan’s right to receive any payments then due under our agreement would be senior to the rights of the holders of our common stock to receive any proceeds from the liquidation of our GPCR program assets.
13
The term of our agreement with Vulcan is 35 years, provided that the term will automatically extend until the cumulative net proceeds that we receive from the GPCR program are approximately $1.5 billion. The term of our agreement with LSDF expires on the six-month anniversary following the last date that we deliver a report related to our incurrence of grant-funded expenses described in the agreement, provided that certain obligations will survive the expiration of the term. The term of our payment obligations to LSDF is the same as that under our agreement with Vulcan.
Competition
Overview. The pharmaceutical and biotechnology industry is highly competitive and characterized by a number of established, large pharmaceutical and biotechnology companies as well as smaller companies like ours. We expect to compete with other pharmaceutical and biotechnology companies, and our competitors may:
| ● | develop and market products that are less expensive, more effective or safer than our future products; |
| ● | commercialize competing products before we can launch our products; |
| ● | operate larger research and development programs, possess greater manufacturing capabilities or have substantially greater financial resources than we do; |
| ● | initiate or withstand substantial price competition more successfully than we can; |
| ● | have greater success in recruiting skilled technical and scientific workers from the limited pool of available talent; |
| ● | more effectively negotiate third-party licenses and strategic relationships; and |
| ● | take advantage of acquisition or other opportunities more readily than we can. |
We expect to compete for market share against large pharmaceutical and biotechnology companies, smaller companies that are collaborating with larger pharmaceutical companies, new companies, academic institutions, government agencies and other public and private research organizations. In addition, the pharmaceutical and biotechnology industry is characterized by rapid technological change. Because our research approach integrates many technologies, it may be difficult for us to remain current with the rapid changes in each technology. Further, our competitors may render our technologies obsolete by advancing their existing technological approaches or developing new or different approaches. If we fail to stay at the forefront of technological change, we may be unable to compete effectively.
Drug Candidates, Development Programs and Platforms. With respect to our development of therapeutics targeting complement-mediated disorders, there are multiple companies developing potential therapies targeting the complement system. Although none of these potential therapies, to our knowledge, selectively inhibit the lectin pathway, there are other companies developing alternative pathway inhibitors. There are also a number of complement-targeted therapeutics that have been approved for commercial use, including Soliris® (eculizumab), Ultomiris® (ravulizumab-cwvz), Empaveli® (pegcetacoplan) and Tavneos® (avocopan), with which narsoplimab and/or OMS906 will compete if either is approved for any indication(s) for which one or more of these products are also approved.
We are aware of other companies attempting to de-orphanize orphan GPCRs. If any of these companies is able to de-orphanize an orphan GPCR before we unlock this receptor, we may be unable to establish an exclusive or commercially valuable intellectual property position around that orphan GPCR.
14
Intellectual Property
We have retained control of all worldwide manufacturing, marketing and distribution rights for each of our drug candidates and programs. Some of our drug candidates and programs are based on inventions and other intellectual property rights that we acquired through assignments, exclusive licenses or acquisitions described in further detail under “License and Development Agreements” below.
As of February 9, 2022, we owned or held worldwide exclusive licenses to a total of 80 issued patents and 56 pending patent applications in the U.S. and 1,205 issued patents and 516 pending patent applications in foreign markets directed to therapeutic compositions and methods related to our research and development programs. For each program, our decision to seek patent protection in specific foreign markets, in addition to the U.S., is based on many factors, including one or more of the following: our available resources, the size of the commercial market, the presence of a potential competitor or a contract manufacturer in the market and whether the legal authorities in the market effectively enforce patent rights.
| ● | MASP-2 Program - Narsoplimab (OMS721). We hold worldwide exclusive licenses to rights in connection with MASP-2, the antibodies targeting MASP-2 and the therapeutic applications for those antibodies from the University of Leicester, MRC and Helion. As of February 9, 2022, we exclusively controlled 29 issued patents and 31 pending patent applications in the U.S., and 632 issued patents and 367 pending patent applications in foreign markets, related to our MASP-2 program. Our MASP-2 and narsoplimab patents have terms that will expire as late as 2037 and, if currently pending patent applications are issued, as late as 2042. |
| ● | MASP-3 Program - OMS906. We own and exclusively control under a license from the University of Leicester all rights to methods of treating various disorders and diseases by inhibiting MASP-3. As of February 9, 2022, we exclusively controlled three issued patents and five pending patent applications in the U.S. and 150 issued and 86 pending patent applications in foreign markets that are related to our MASP-3 program. |
| ● | PPARγ Program - OMS405. As of February 9, 2022, we owned three issued patents and one pending patent application in the U.S., and 37 issued patents and 7 pending patent applications in foreign markets, directed to our discoveries linking PPARγ and addictive disorders. |
| ● | PDE7 Program - OMS527. As of February 9, 2022, we owned two issued patents and one pending patent application in the U.S., and 61 issued patents and twoe pending patent applications in foreign markets directed to our discoveries linking PDE7 to movement disorders, as well as two issued patent and two pending patent applications in the U.S., and 49 issued patents and 11 pending patent applications in foreign markets directed to the link between PDE7 and addiction and compulsive disorders. Additionally, under a license from Daiichi Sankyo, we exclusively control rights to three issued U.S. patents and 62 issued patents in foreign markets that are directed to proprietary PDE7 inhibitors. For a more detailed description of our agreement with Daiichi Sankyo, see “License and Development Agreements” below. |
| ● | GPCR Platform. As of February 9, 2022, we owned seven issued patents and 12 pending patent applications in the U.S., and 56 issued patents and 24 pending patent applications in foreign markets, which are directed to previously unknown links between specific molecular targets in the brain and a series of CNS disorders, to our CRA and to other research tools that are used in our GPCR program, and to orphan GPCRs and other GPCRs for which we have identified functionally interacting compounds using our CRA. Two of the pending patent applications in the U.S. and the 24 pending patent applications in foreign markets are directed to GPR174. |
All of our employees enter into our standard employee proprietary information and inventions agreement, which includes confidentiality provisions and provides us ownership of all inventions and other intellectual property made by our employees that pertain to our business or that relate to our employees’ work for us or that result from the use of our resources. Our commercial success will depend in part on obtaining and maintaining patent protection and trade secret protection of the use, formulation and structure of our drug candidates and the methods used to manufacture them, as well as on our ability to defend successfully these patents against third-party challenges. Our ability to protect our drug
15
candidates from unauthorized making, using, selling, offering to sell or importing by third parties is dependent on the extent to which we have rights under valid and enforceable patents that cover these activities.
The patent positions of pharmaceutical, biotechnology and other life sciences companies can be highly uncertain and involve complex legal and factual questions for which important legal principles remain unresolved. No consistent policy regarding the breadth of claims allowed in biotechnology patents has emerged to date in the U.S., and tests used for determining the patentability of patent claims in all technologies are in flux. The pharmaceutical, biotechnology and other life sciences patent situation outside the U.S. is even more uncertain. Changes in either the patent laws or in interpretations of patent laws in the U.S. and other countries may diminish the value of our intellectual property. Accordingly, we cannot predict the breadth of claims that may be allowed or enforced in the patents that we own or have licensed or in third-party patents.
We have registered, and intend to maintain, the trademark “OMEROS” within the U.S. Patent and Trademark Office in connection with the products and services we offer. We are not aware of any material claims of infringement or other challenges to our right to use the “OMEROS” trademark in the U.S.
Government Regulation
Government authorities in the U.S., the EU and other countries extensively regulate the research, development, testing, manufacture, labeling, promotion, advertising, distribution, marketing, and export and import of drug and biologic products including the drug candidates that we are developing. Failure to comply with applicable requirements, both before and after receipt of regulatory approval, may subject us, our third-party manufacturers, and other partners to administrative and judicial sanctions, such as warning letters, product recalls, product seizures, a delay in approving or refusal to approve pending applications, civil and other monetary penalties, total or partial suspension of production or distribution, injunctions, and/or criminal prosecutions.
In the U.S., our drug candidates are regulated by FDA as drugs or biologics under the FDCA and implementing regulations and under the Public Health Service Act (“PHSA”). In the EU, our drug candidates are regulated by the EMA and national medicines regulators under the rules governing medicinal products in the EU as well as national regulations in individual countries. The marketing authorization for OMIDRIA in the U.S. has been transferred to Rayner and the marketing authorizations in the EU and United Kingdom are in process of being transferred to Rayner, as required by the Asset Purchase Agreement pursuant to which Omeros divested OMIDRIA and related assets, including such marketing approvals, in December 2021. Our drug candidates are in various stages of testing and none of our drug candidates has received marketing approval from FDA or the applicable regulatory authorities in the EU.
The steps required before a product may be approved for marketing by FDA, or the applicable regulatory authorities outside of the U.S., typically include the following:
| ● | formulation development and manufacturing process development; |
| ● | preclinical laboratory and animal testing; |
| ● | submission to FDA of an Investigational New Drug application (“IND”) for human clinical testing, which must become effective before human clinical trials may begin; and in countries outside the U.S., a Clinical Trial Application (“CTA”), is filed according to the country’s local regulations; |
| ● | adequate and well-controlled human clinical trials to establish the efficacy and safety of the product for each indication for which approval is sought; |
| ● | adequate assessment of drug product stability to determine shelf life/expiry dating; |
16
| ● | in the U.S., submission to FDA of a New Drug Application (“NDA”), in the case of a drug product, or a BLA in the case of a biologic product and, in Europe, submission to the EMA or a national regulatory authority of an MAA; |
| ● | satisfactory completion of inspections of one or more clinical sites at which clinical trials with the product were carried out and of the manufacturing facility or facilities at which the product is produced to assess compliance with Good Clinical Practices (“GCPs”), and cGMPs; and |
| ● | FDA review and approval of an NDA or BLA, or review and approval of an MAA by the applicable regulatory authorities in the EU. |
Manufacturing. Manufacturing of drug products for use in clinical trials must be conducted according to relevant national and international guidelines, for example, cGMP. Process and formulation development are undertaken to design suitable routes to manufacture the drug substance and the drug product for administration to animals or humans. Analytical development is undertaken to obtain methods to quantify the potency, purity and stability of the drug substance and drug product as well as to measure the amount of the drug substance and its metabolites in biological fluids, such as blood.
Preclinical Tests. Preclinical tests include laboratory evaluations and animal studies to assess efficacy, toxicity and pharmacokinetics. The results of the preclinical tests, together with manufacturing information, analytical data, clinical development plan, and other available information are submitted as part of an IND or CTA.
The IND/CTA Process. An IND or CTA must become effective before human clinical trials may begin. INDs are extensive submissions including, among other things, the results of the preclinical tests, together with manufacturing information and analytical data. In addition to including the results of the preclinical studies, the IND will also include one or more protocols for proposed clinical trials detailing, among other things, the objectives of the clinical trial, the parameters to be used in monitoring safety and the effectiveness criteria to be evaluated. An IND will become effective 30 days after receipt by FDA unless, before that time, FDA raises concerns or questions and imposes a clinical hold. In that event, the IND sponsor and FDA must resolve any outstanding FDA concerns or questions before the clinical hold is lifted and clinical trials can proceed. Similarly, a CTA must be cleared by the local independent ethics committee and competent authority prior to conducting a clinical trial in the country in which it was submitted. There can be no assurance that submission of an IND or CTA will result in authorization to commence clinical trials. Once an IND or CTA is in effect, there are certain reporting requirements.
Clinical Trials. Clinical trials involve the administration of the investigational product to human subjects under the supervision of qualified personnel and must be conducted in accordance with local regulations and GCPs. Clinical trials are conducted under protocols detailing, for example, the parameters to be used in monitoring patient safety and the efficacy criteria, or endpoints, to be evaluated. Each trial must be reviewed and approved by an independent institutional review board or ethics committee for each clinical site at which the trial will be conducted before it can begin. Clinical trials are typically conducted in three defined phases, but the phases may overlap or be combined:
| ● | Phase 1 usually involves the initial administration of the investigational product to human subjects, who may or may not have the disease or condition for which the product is being developed, to evaluate the safety, dosage tolerance, pharmacodynamics and, if possible, to gain an early indication of the effectiveness of the product. |
| ● | Phase 2 usually involves trials in a limited patient population with the disease or condition for which the product is being developed to evaluate appropriate dosage, to identify possible adverse side effects and safety risks, and to evaluate preliminarily the effectiveness of the product for specific indications. |
| ● | Phase 3 clinical trials usually further evaluate and confirm effectiveness and test further for safety by administering the product in its final form in an expanded patient population. |
17
We, our product development partners, institutional review boards or ethics committees, FDA or other regulatory authorities may suspend or terminate clinical trials at any time on various grounds, including a belief that the subjects are being exposed to an unacceptable health risk.
Disclosure of Clinical Trial Information. Sponsors of clinical trials of certain FDA-regulated products, including prescription drugs, are required to register and disclose certain clinical trial information on a public website maintained by the U.S. National Institutes of Health. Information related to the product, patient population, phase of investigation, study sites and investigator, and other aspects of the clinical trial is made public as part of the registration. Sponsors are also obligated to disclose the results of these trials after completion. Disclosure of the results of these trials can be delayed for up to two years if the sponsor certifies that it is seeking approval of an unapproved product or that it will file an application for approval of a new indication for an approved product within one year. Clinical trials conducted in European countries are required to be registered at a similar public database maintained and overseen by European health authorities. Competitors may use this publicly available information to gain knowledge regarding the design and progress of our development programs.
The Application Process. If the necessary clinical trials are successfully completed, the results of the preclinical trials and the clinical trials, together with other detailed information, including information on the manufacture and composition of the product, are submitted to FDA in the form of an NDA or a BLA, as applicable, and to the EMA or national regulators in the form of an MAA, requesting approval to market the product for a specified indication. In the EU, an MAA may be submitted to the EMA for review and, if the EMA gives a positive opinion, the EC may grant a marketing authorization that is valid across the EU (centralized procedure). Alternatively, an MAA may be submitted to one or more national regulators in the EU according to one of several national or decentralized procedures. The type of submission in Europe depends on various factors and must be cleared by the appropriate authority prior to submission. For most of our drug candidates, the centralized procedure will be either mandatory or available as an option.
If the regulatory authority determines that the application is not acceptable, it may refuse to accept the application for filing and review, outlining the deficiencies in the application and specifying additional information needed to file the application. Notwithstanding the submission of any requested additional testing or information, the regulatory authority ultimately may decide that the proposed product is not safe or effective, or that the application does not otherwise satisfy the criteria for approval. In the U.S., to support an approval an NDA must demonstrate, among other things, that the proposed drug product is safe and effective, has a favorable benefit-risk profile, is manufactured in a way that preserves its identity, strength, purity and potency, and that its labeling is adequate and not false or misleading. A similar standard exists for BLAs. Before approving an NDA or BLA, or an MAA, FDA or the EMA, respectively, may inspect one or more of the clinical sites at which the clinical studies were conducted to ensure that GCPs were followed and may inspect facilities at which the product is manufactured to ensure satisfactory compliance with cGMP. The FDA may refer the NDA or BLA to an advisory committee for review and recommendation as to whether the application should be approved and under what conditions. The FDA is not bound by the recommendation of an advisory committee, but it generally follows such recommendation. In addition, even if a drug candidate satisfied its endpoints with statistical significance during clinical trials, FDA could determine that the overall balance of risks and benefits for the drug candidate is not adequate to support approval, or only justifies approval for a narrow set of clinical uses and/or subject to restricted distribution or other burdensome post-approval requirements or limitations. If approval is obtained changes to the approved product such as adding new indications, manufacturing changes, or additional labeling claims will require submission of a supplemental application, referred to as a variation in the EU, or, in some instances, a new application, for further review and approval. The testing and approval process requires substantial time, effort, and financial resources, and we cannot be sure that any future approval will be granted on a timely basis, if at all.
Some of our drug candidates, such as those from our MASP-2 and MASP-3 programs, are considered biologics because they are derived from natural sources as opposed to being chemically synthesized. The added complexity associated with manufacturing biologics may result in additional monitoring of the manufacturing process and product changes.
In addition, we, our suppliers and our contract manufacturers are required to comply with extensive regulatory requirements both before and after approval. For example, we must establish a pharmacovigilance system and are required to report adverse reactions and production problems, if any, to the regulatory authorities. If any of our drug
18
candidates are approved, we will be required to also comply with certain requirements concerning advertising and promotion for our products. The regulatory authorities may impose specific obligations as a condition of the marketing authorization, such as additional safety monitoring, or the conduct of additional clinical trials or post-marketing safety studies, or the imposition of a Risk Evaluation and Mitigation Strategy (“REMS”), which could include significant restrictions on distribution or use of the product. Also, quality control and manufacturing procedures must continue to conform to cGMPs after approval. Accordingly, manufacturers must continue to expend time, money, and effort in all areas of regulatory compliance, including production and quality control to comply with cGMPs. In addition, discovery of problems such as safety issues may result in changes in labeling or restrictions on a product manufacturer or marketing authorization holder, including removal of the product from the market.
Fast-Track and Priority Review Designations. Section 506(b) of the FDCA provides for the designation of a drug as a fast-track product if it is intended, whether alone or in combination with one or more other drugs, for the treatment of a serious or life-threatening disease or condition, and it demonstrates the potential to address unmet medical needs for such a disease or condition. A program with fast-track status is afforded greater access to FDA for the purpose of expediting the product’s development, review and potential approval. Many products that receive fast-track designation are also considered appropriate to receive priority review, and their respective applications may be accepted by FDA as a rolling submission in which portions of an NDA or BLA are reviewed before the complete application is submitted. Together, these may reduce time of development and FDA review time. In Europe, products that are considered to be of major public health interest are eligible for accelerated assessment, which shortens the review period. The grant of fast-track status, priority review or accelerated assessment does not alter the standard regulatory requirements for obtaining marketing approval.
Breakthrough Therapy Designation. In 2012, Congress enacted the Food and Drug Administration Safety and Innovation Act. This law established a regulatory process allowing for increased interactions with FDA with the goal of expediting development and review of products designated as “breakthrough therapies.” A product may be designated as a breakthrough therapy if it is intended, either alone or in combination with one or more other products, to treat a serious or life-threatening disease or condition and preliminary clinical evidence indicates that the product may demonstrate substantial improvement over existing therapies on one or more clinically significant endpoints, such as substantial treatment effects observed early in clinical development. The FDA may take certain actions with respect to breakthrough therapies, including holding meetings with the sponsor throughout the development process; providing timely advice to the product sponsor regarding development and approval; involving more senior staff in the review process; assigning a cross-disciplinary project lead for the review team; and taking other steps to design the clinical trials in an efficient manner.
Accelerated Approval. The FDA may grant accelerated approval to a product for a serious or life-threatening condition that provides a meaningful therapeutic advantage to patients over existing treatments based upon a determination that the product has an effect on a surrogate endpoint that is reasonably likely to predict clinical benefit. The FDA may also grant accelerated approval for such a condition when the product has an effect on an intermediate clinical endpoint that can be measured earlier than an effect on irreversible morbidity or mortality and that is reasonably likely to predict an effect on irreversible morbidity or mortality or other clinical benefit. In both cases, FDA must take into account the severity, rarity, or prevalence of the condition and the availability or lack of alternative treatments. Studies that are conducted to demonstrate a drug’s effect on a surrogate or intermediate clinical endpoint for accelerated approval must be adequate and well-controlled as required by the FDCA.
Following accelerated approval, FDA requires that the company provide confirmatory evidence, which may include certain adequate and well-controlled post-marketing clinical studies to verify the clinical benefit of the product, and FDA may impose restrictions on distribution to assure safe use. Confirmatory studies are typically required to be underway at the time of the accelerated approval. If the required confirmatory studies fail to verify the clinical benefit of the drug, or if the applicant fails to perform the required confirmatory studies with due diligence, FDA may withdraw approval of the drug under streamlined procedures in accordance with the Agency’s regulations. The Agency may also withdraw approval of a drug if, among other things, other evidence demonstrates that the drug product is not shown to be safe or effective under its conditions of use.
19
The EU also has accelerated approval programs. In the EU, a marketing authorization may be granted on the basis of less complete data than are normally required in certain “exceptional circumstances,” such as when the product’s indication is encountered so rarely that the applicant cannot reasonably be expected to provide comprehensive data. Alternatively, a conditional marketing authorization may be granted prior to obtaining the comprehensive clinical data required for a full MAA if a product fulfills an unmet medical need and the benefit to public health of the product’s immediate availability outweighs the risk inherent in the incomplete data.
Orphan Drug Designation. Under the Orphan Drug Act (“ODA”), FDA may grant orphan drug designation to drugs or biologics intended to treat a rare disease or condition that affects fewer than 200,000 individuals in the U.S. or more than 200,000 individuals in the U.S. for which the cost of developing and making the product available in the U.S. for this type of disease or condition is not likely to be recovered from U.S. sales for that product. The granting of orphan designation does not alter the standard regulatory requirements (other than payment of certain fees and the applicability of certain pediatric assessment requirements), nor does it alter the standards or process for obtaining marketing approval. The sponsor of a product that has an orphan drug designation qualifies for various development incentives specified in the ODA, including a tax credit of up to 25% of expenditures on qualified clinical testing for the orphan drug. Furthermore, if the orphan designated product subsequently receives the first FDA approval for the orphan indication, the product is entitled to an orphan drug exclusivity period, which means that FDA may not grant approval to any other application to market the same drug for the same indication for a period of seven years except in limited circumstances, such as a showing of clinical superiority to the product with orphan drug exclusivity for the protected indication. Orphan drug exclusivity does not prevent FDA from approving a different drug for the same disease or condition, or the same drug for a different disease or condition. The EU has a similar Orphan Drug program to that of the U.S., and it is administered through the EMA’s Committee for Orphan Medicinal Products.
Pediatric Testing and Exclusivity. In the U.S., NDAs and BLAs are subject to both mandatory pediatric testing requirements and voluntary pediatric testing incentives in the form of exclusivity. An additional six months of exclusivity in the U.S. may be granted to a sponsor of an NDA or BLA if the sponsor conducts certain pediatric studies, which studies are conducted pursuant to a written request from FDA. This process is initiated when FDA issues a Written Request for pediatric studies to determine if the drug or biologic could have meaningful pediatric health benefits. If FDA determines that the sponsor has conducted the requested pediatric studies in accordance with the written request, then an additional six months of exclusivity may attach in the case of a drug to any other regulatory exclusivity or patent protection applicable to the drug and, in the case of a biologic, to any other regulatory exclusivity applicable to the biologic. The EU has a similar requirement and incentive for the conduct of pediatric studies according to the pediatric investigation plan, which must be adopted by the EMA before an MAA may be submitted.
Expanded Access. “Expanded access” refers to the use of an investigational drug where the primary purpose is to diagnose, monitor, or treat a patient’s disease or condition rather than to collect information about the safety or effectiveness of a drug. There are three FDA-recognized categories of expanded access trials: expanded access for individual patients, including for emergency use; expanded access for intermediate-size patient populations; and expanded access for large patient populations under a treatment IND or treatment protocol. For all types of expanded access, FDA must determine prior to authorizing expanded access that: (1) the patient or patients to be treated have a serious or life-threatening disease or condition and there is no comparable or satisfactory alternative therapy; (2) the potential patient benefit justifies the potential risks of use and that the potential risks are not unreasonable in the context of the disease or condition to be treated; and (3) granting the expanded access will not interfere with the initiation, conduct, or completion of clinical studies in support of the drug’s approval. Only a licensed physician or the drug’s manufacturer may apply for expanded access. Manufacturers are not required to supply the investigational product for expanded access. The FDA has established streamlined processes for physicians to request individual patient expanded access whereby physicians can submit a single patient IND. In cases of individual patient emergency expanded access, physicians can receive FDA approval for access by phone and follow up with the abbreviated form. In addition, the sponsor of an expanded access IND must submit IND safety reports and, in the cases of protocols continuing for one year or longer, annual reports to FDA.
U.S. Labeling, Marketing and Promotion. The FDA closely regulates the labeling, marketing and promotion of drugs. In general, our labeling and promotion must not be false or misleading in any particular, and claims that we make must be adequately substantiated. In addition, our approved labeling must include adequate directions to physicians for
20
each intended use of our products. Failure to comply with these requirements can result in adverse publicity, warning letters, corrective advertising, injunctions and potential civil and criminal penalties.
In addition to regulation by FDA, the research, manufacturing, distribution, sale and promotion of drug products in the U.S. are subject to regulation by various federal, state and local authorities, including CMS, other divisions of the U.S. Department of Health and Human Services (e.g., the Office of Inspector General), the U.S. Department of Justice, state Attorneys General, and other state and local government agencies. All of these activities are also potentially subject to federal and state consumer protection and unfair competition laws. Violations of these laws are punishable by prison sentences, criminal fines, administrative civil money penalties, and exclusion from participation in federal healthcare programs.
There are also an increasing number of state laws that require manufacturers to make reports to states on pricing and marketing information or impose other special requirements for the sale and marketing of drug products. Many of these laws contain ambiguities as to what is required to comply with the laws. In addition, federal and state “transparency laws” require manufacturers to track and report certain payments made to health care providers and, under some state laws, other information concerning our products. These laws may affect our sales, marketing and other promotional activities by imposing administrative and compliance burdens on us. In addition, our reporting actions could be subject to the penalty provisions of the pertinent state and federal authorities.
Drug Supply Chain Security Act. Title II (the Drug Supply Chain Security Act (the “DSCSA”)), of the Drug Quality and Security Act imposes on manufacturers of certain pharmaceutical products new obligations related to product tracking and tracing, among others, which began a several-year phase-in process in 2015. Among the requirements of this legislation, manufacturers subject to the DSCSA are required to provide certain documentation regarding the drug product to trading partners to which product ownership is transferred, label drug product with a product identifier (i.e., serialize), respond to verification requests from trading partners, provide transaction documentation upon request by federal or state government entities, and keep certain records regarding the drug product. The transfer of information to subsequent product owners by manufacturers must be done electronically. For products and transactions falling within DSCSA’s scope, manufacturers are required to verify that purchasers of the manufacturers’ products are appropriately licensed. Further, under the DSCSA, covered manufacturers have drug product investigation, quarantine, disposition, and notification responsibilities for product that is reasonably believed or that credible evidence shows to be counterfeit, diverted, stolen, intentionally adulterated such that the product would result in serious adverse health consequences or death, the subject of fraudulent transactions or otherwise unfit for distribution such that they would be reasonably likely to result in serious health consequences or death. Anti-counterfeiting and serialization requirements similar to those under the DSCSA have also been adopted in the EU and became effective in February 2019.
Foreign Regulatory Requirements. Outside of the U.S., our ability to conduct clinical trials or market our products will also depend on receiving the requisite authorizations from the appropriate regulatory authorities. The foreign regulatory approval processes include similar requirements and many of the risks associated with FDA and/or the EU approval process described above, although the precise requirements may vary from country to country. In the EU, once an MAA is granted, the product must be “placed on the market” in at least one EEA country within three years of the date of authorization. “Placed on the market” is defined as when the medicinal product is “released into the distribution chain,” i.e., out of the direct control of the marketing authorization holder. In July 2021, we placed OMIDRIA on the market in the EU, on a limited basis, which maintained the ongoing validity of the European marketing authorization for OMIDRIA. The EU marketing authorization is in the process of being transferred to Rayner as required under the Asset Purchase Agreement pursuant to which we divested OMIDRIA and related assets to Rayner in December 2021.
Hatch-Waxman Act. In seeking approval for a drug through an NDA, applicants are required to list with FDA each patent with claims that cover the applicant’s drug or an approved method of use of the drug. Upon approval of a drug, each of the patents listed in the application for the drug is then published in FDA’s Approved Drug Products with Therapeutic Equivalence Evaluations, commonly known as the Orange Book. Drugs listed in the Orange Book can, in turn, be cited by potential competitors in support of approval of an ANDA or a 505(b)(2) application. In this case the original NDA, i.e., the pioneer drug, is known as the “listed” drug or “reference-listed” drug. An ANDA provides for marketing of a drug that has the same active ingredients and, in some cases (e.g., ophthalmology), also the same inactive ingredients, in the same strengths, route of administration and dosage form as the listed drug and has been shown
21
through testing to be bioequivalent to the listed drug or receives a waiver from bioequivalence testing. ANDA applicants are generally not required to conduct or submit results of preclinical or clinical tests to prove the safety or effectiveness of their drug, other than the requirement for bioequivalence testing. Drugs approved in this way are considered therapeutically equivalent, and are commonly referred to as “generic equivalents” to the listed drug. These drugs then generally can be substituted by pharmacists under prescriptions written for the original listed drug.
The ANDA or 505(b)(2) applicant is required to certify to FDA concerning any patents listed for the referenced approved drug in FDA’s Orange Book. Specifically, for each listed patent, the applicant must certify that: (1) the required patent information has not been filed; (2) the listed patent has expired; (3) the listed patent has not expired but will expire on a particular date and approval is sought after patent expiration; or (4) the listed patent is invalid, unenforceable or will not be infringed by the new drug. A certification that the new drug will not infringe the already approved drug’s listed patents or that such patents are invalid or unenforceable is called a Paragraph IV certification. If the ANDA or 505(b)(2) applicant does not include a Paragraph IV certification, the ANDA or 505(b)(2) application will not be approved until all of the listed patents claiming the referenced drug have expired, except for any listed patents that only apply to uses of the drug not being sought by the ANDA or 505(b)(2) applicant.
If the ANDA or 505(b)(2) applicant has made a Paragraph IV certification, the applicant must also send notice of a Paragraph IV Notice Letter to the NDA and patent holders once the ANDA or 505(b)(2) application has been accepted for filing by FDA. The NDA and patent holders may then initiate a patent infringement lawsuit in response to the notice of the Paragraph IV Notice Letter. The filing of a patent infringement lawsuit within 45 days of the receipt of notice of a Paragraph IV Notice Letter automatically prevents FDA from approving the ANDA until the earlier of 30 months, expiration of the patent, settlement of the lawsuit, modification by a court or a decision in the infringement case that is favorable to the ANDA or 505(b)(2) applicant.
The ANDA or 505(b)(2) application also will not be approved until any applicable non-patent exclusivity, such as exclusivity for obtaining approval of a new chemical entity, listed in the Orange Book for the reference-listed drug has expired. The U.S. Drug Price Competition and Patent Term Restoration Act of 1984, more commonly known as the Hatch-Waxman Act, provides a period of five years following approval of a drug containing no previously approved active moiety, during which ANDAs for generic versions of those drugs and 505(b)(2) applications referencing those drugs cannot be submitted unless the submission contains a Paragraph IV challenge to a listed patent, in which case the submission may be made four years following the original drug approval. The Hatch-Waxman Act also provides for a period of three years of exclusivity following approval of a listed drug that contains previously approved active ingredients but is approved in a new dosage form, route of administration or combination, or for a new use, the approval of which was supported by new clinical trials other than bioavailability studies that were essential to the approval and conducted by or for the sponsor. During those three years of exclusivity, FDA cannot grant approval of an ANDA or 505(b)(2) application for the protected dosage form, route of administration or combination, or use of that listed drug.
In December 2019, a piece of legislation referred to as the Creating and Restoring Equal Access to Equivalent Samples Act of 2019 (“CREATES Act”) was signed into law, which is intended to address the concern that some brand manufacturers have improperly denied generic and biosimilar product developers access to samples of brand products. The CREATES Act establishes a private cause of action that permits a generic or biosimilar product developer to sue the brand manufacturer to compel it to furnish the necessary samples on commercially reasonable, market-based terms. If the developer prevails, the court may grant the developer a monetary award up to the brand product’s revenue for the period of delay in providing samples.
Biosimilars. The enactment of federal healthcare reform legislation in March 2010 provided a new pathway for approval of follow-on biologics (i.e., biosimilars) under the PHSA. FDA licensure of a biosimilar is dependent upon many factors, including a showing that the proposed biosimilar is “highly similar” to the reference product, notwithstanding minor differences in clinically inactive components, and has no clinically meaningful differences from the reference product in terms of safety, purity, and potency. The types of data ordinarily required in a biosimilar application to show high similarity include analytical data, animal studies (including toxicity studies), and clinical studies (including immunogenicity and pharmacokinetic/pharmacodynamic studies). A biosimilar must seek licensure for a condition of use for which the reference-listed product is licensed.
22
Furthermore, the PHSA provides that for a biosimilar to be considered “interchangeable” (i.e., the biological product may be substituted for the reference product without the intervention of the health care provider who prescribed the reference product), the applicant must make an additional showing that the biosimilar can be expected to produce the same clinical result as the reference product in any given patient, and if the product is administered more than once to a patient, that risks in terms of safety or diminished efficacy of alternating or switching between the biological product and the reference product is no greater than the risk of using the reference product without switching. Although FDA has provided guidance on what information and data an applicant should submit to enable an interchangeability determination, thus far FDA has not licensed any biologic as being interchangeable with its reference product.
The PHSA also provides a period of exclusivity for pioneer biologics. Specifically, FDA may not accept a biosimilar application referencing data from a pioneer biologic (i.e., one approved through a full BLA) until four years have elapsed from the date of first licensure of the pioneer biologic. FDA may not approve a biosimilar application referencing data from a pioneer biologic until 12 years have elapsed since the date of first licensure of the pioneer biologic. There are certain restrictions and limitations on the types of BLAs that are eligible for biologics exclusivity as well as what constitutes the date of first licensure for a pioneer biologic.
In the EU, a pathway for the approval of biosimilars has existed since 2005.
Healthcare compliance laws. In the U.S., commercialization of our drug candidates, if approved, is subject to regulation and enforcement under a number of federal and state healthcare compliance laws administered and enforced by various agencies. These include, but are not limited to, the following:
| ● | the federal Anti-Kickback Statute, which prohibits offering or paying anything of value to a person or entity to induce or reward referrals for goods or services reimbursed by a federal healthcare program such as Medicare or Medicaid; |
| ● | the federal False Claims Act, which prohibits presenting or causing to be presented a false claim for payment by a federal healthcare program, and which has been interpreted to also include claims caused by improper drug-manufacturer product promotion or the payment of kickbacks; |
| ● | a variety of governmental pricing, price reporting, and rebate requirements, including those under Medicaid and the Veterans Health Care Act; and |
| ● | the so-called Sunshine Act and certain provisions of the Affordable Care Act, which require that we report to the federal government information on certain financial payments and other transfers of value made to certain health care providers and institutions, as well as certain information regarding our distribution of drug samples. |
In addition to these federal law requirements, several U.S. states have enacted similar laws requiring periodic reporting and/or disclosure related to our marketing, sales and other activities, or regulating certain sales and marketing activities, such as provision of meals, gifts or entertainment to certain health care providers. We may also be subject to federal or state privacy laws if we receive protected patient health information.
Similar requirements apply to our operations outside of the U.S. Laws in the U.S. such as the Foreign Corrupt Practices Act prohibit the offering or payment of bribes or inducements to foreign public officials for business, including physicians or other medical professionals who are employees of public healthcare entities. In addition, many non-U.S. jurisdictions in which we operate, or may operate in the future, have their own laws similar to the healthcare compliance laws that exist in the U.S.
Pharmaceutical Pricing and Reimbursement
Overview. In both U.S. and foreign markets, our ability to commercialize our drug candidates successfully, and to attract commercialization partners for our drug candidates, depends in significant part on the availability of adequate financial coverage and reimbursement from third-party payers including, in the U.S., managed care organizations and other private health insurers as well as governmental payers such as the Medicare and Medicaid programs.
23
Reimbursement by a third-party payer may depend on a number of factors, including the payer’s determination that use of a product is:
| ● | a covered benefit under its health plan; |
| ● | safe, effective and medically necessary; |
| ● | appropriate for the specific patient; |
| ● | cost-effective; and |
| ● | neither experimental nor investigational. |
Reimbursement by government payers is based on statutory authorizations and complex regulations that may change with annual or more frequent rulemaking, as well as legislative reform measures.
Third-party private and governmental payers are increasingly challenging the prices charged for medicines and examining their cost-effectiveness in addition to their safety and efficacy. We may need to conduct expensive pharmacoeconomic studies in order to demonstrate the cost effectiveness of our products or drug candidates. Even with the availability of such studies, third-party private and/or governmental payers may not provide coverage and reimbursement for our drug candidates, in whole or in part.
United States. Political, economic and regulatory influences are subjecting the healthcare industry in the U.S. to fundamental changes. There have been, and we expect there will continue to be, legislative and regulatory proposals to change the healthcare system in ways that could significantly affect our business. For example, the 2010 Affordable Care Act (the “ACA”), is intended to broaden access to health insurance, reduce or constrain the growth of healthcare spending, enhance remedies against fraud and abuse, add transparency requirements for the healthcare and health insurance industries, impose new taxes and fees on the health industry and impose additional health policy reforms. Other legislative changes included a two percent across-the-board reduction to Medicare payments to providers, effective April 1, 2013, which, due to subsequent legislative amendments, will stay in effect through fiscal year 2029 unless additional congressional action is taken. (A temporary suspension of this reduction during the public health emergency for the pandemic is currently scheduled to expire on March 31, 2022.) The American Taxpayer Relief Act of 2012, among other things, reduced Medicare payments to several providers, and increased the period for the government to recover overpayments to providers from three to five years. In December 2017, portions of the ACA dealing with the individual mandate insurance requirement were effectively repealed by the Tax Cuts and Jobs Act of 2017. The latest court challenge to the ACA failed in June 2021, when the United States Supreme Court held that the individual plaintiffs and states lacked standing to challenge the constitutionality of the ACA.
In November 2020, CMS issued an interim final rule through the CMS Innovation Center whereby Medicare Part B reimbursement for “certain high-cost prescriptions drugs” would be no more than most-favored-nation price (i.e., the lowest price) after adjustments, for a pharmaceutical product that the drug manufacturer sells in a member country of the Organization for Economic Cooperation and Development that has a comparable per-capita gross domestic product. In December 2020, the U.S. District Court in Northern California issued a nationwide preliminary injunction against implementation of the interim final rule. The Biden administration subsequently withdrew the interim final rule. The Biden administration has indicated that lowering prescription drug prices is a priority, but it is not yet clear what steps the administration will take or whether such steps will be successful. We cannot predict the ultimate content, timing or effect of any healthcare reform legislation or executive order or the impact that the resulting changes may have on us.
We are unable to predict what additional legislation, regulations, policies or court orders, if any, relating to the healthcare industry or coverage and reimbursement may be enacted or imposed in the future or what effect such legislation, regulations, policies or court orders would have on our business. Any cost-containment measures, including those listed above, or other healthcare system reforms that are adopted could have a material adverse effect on our business prospects and financial operations.
24
Europe. Governments in the various member states of the EU influence or control the price of medicinal products in their countries through their pricing and reimbursement rules and control of national healthcare systems that fund a large part of the cost of those products to consumers. To obtain reimbursement or pricing approval, some of these countries may require the completion of clinical trials or pharmacoeconomic studies that assess the cost-effectiveness of a product or drug candidate relative to currently available therapies or relative to a specified standard. The downward pressure on healthcare costs in general, and prescription medicines in particular, has become very intense and is creating increasingly high barriers to the entry of new products in these markets.
Research and Development
We have built a research and development organization that includes expertise in discovery research, preclinical development, product formulation, analytical and medicinal chemistry, manufacturing, clinical development and regulatory and quality assurance. We operate cross-functionally and are led by an experienced management team. We use rigorous project management techniques to make disciplined strategic decisions regarding our research and development programs and to limit the risk profile of our product pipeline. We also access relevant market information and key opinion leaders in creating target product profiles and, when appropriate, as we advance our programs to commercialization. We engage third parties on a limited basis to conduct portions of our preclinical research; however, we are not substantially dependent on any third parties for our preclinical research nor do any of these third parties conduct a major portion of our preclinical research. We also engage multiple clinical sites to conduct our clinical trials. None of these sites conduct the major portion of our clinical trials and we are not substantially dependent on any one of them.
Employees
As of December 31, 2021, we had 213 full-time employees, 135 of whom are in research and development, 24 of whom are in sales and marketing and 54 of whom are in finance, legal, business development and administration. Our full-time employees include five with M.D.s and 39 with Ph.Ds., of whom one and 23, respectively, are in research and development. None of our employees are represented by a labor union, and we consider our employee relations to be good.
Information about Our Executive Officers and Significant Employees
The following table provides information regarding our executive officers and significant employees as of March 1, 2022:
Name |
| Age |
| Position(s) |
Executive Officers: | ||||
Gregory A. Demopulos, M.D. | 63 | President, Chief Executive Officer and Chairman of the Board of Directors | ||
Michael A. Jacobsen | 63 | Vice President, Finance, Chief Accounting Officer and Treasurer | ||
Peter B. Cancelmo, J.D. | 43 | Vice President, General Counsel and Secretary | ||
Significant Employees: | ||||
Christopher S. Bral, Ph.D. | 56 | Vice President, Nonclinical Development | ||
Nadia Dac | 52 | Vice President, Chief Commercial Officer | ||
George A. Gaitanaris, M.D., Ph.D. | 65 | Vice President, Science and Chief Scientific Officer | ||
Bruce Meiklejohn, Ph.D. | 62 | Vice President, Chemistry, Manufacturing and Controls | ||
Catherine A. Melfi, Ph.D. | 62 | Vice President, Regulatory Affairs & Quality Systems and Chief Regulatory Officer | ||
Tina Quinton, J.D., M.S. | 59 | Vice President, Patents | ||
J. Steven Whitaker, M.D., J.D. | 66 | Vice President, Chief Medical Officer | ||
Peter W. Williams | 54 | Vice President, Human Resources |
Gregory A. Demopulos, M.D. founded our company and has served as our president, chief executive officer and chairman of the board of directors since June 1994. He also served as our chief financial officer and treasurer from
25
January 2009 to October 2013 in an interim capacity and as our chief medical officer from June 1994 to March 2010. Prior to founding Omeros, Dr. Demopulos completed his residency in orthopedic surgery at Stanford University and his fellowship training in hand and microvascular surgery at Duke University. Dr. Demopulos currently serves on the board of trustees of the Smead Funds Trust, an open-end mutual fund company registered under the Investment Company Act of 1940. Dr. Demopulos received his M.D. from the Stanford University School of Medicine and his B.S. from Stanford University. Dr. Demopulos is the brother of Peter A. Demopulos, M.D., a member of our board of directors.
Michael A. Jacobsen has served as our vice president, finance, chief accounting officer and treasurer since October 2013. Prior to joining Omeros, Mr. Jacobsen served as vice president of finance of Sarepta Therapeutics, Inc. from September 2011 to May 2013 and as its chief accounting officer from September 2011 to December 2012. From April 2007 to August 2011, Mr. Jacobsen was vice president and chief accounting officer at ZymoGenetics, Inc. Prior to his service with ZymoGenetics, Mr. Jacobsen held various roles at ICOS Corporation, including senior director of finance and corporate controller. From April 1995 to October 2001, Mr. Jacobsen held vice president of finance or chief financial officer roles at three companies in the software, computer hardware and internet retailing industries, two of which were publicly traded. Mr. Jacobsen is a certified public accountant and received his bachelor’s degree in accounting from Idaho State University.
Peter B. Cancelmo, J.D. has served as our vice president, general counsel and secretary since June 2019. He joined Omeros as deputy general counsel, corporate governance and securities in January 2019. Prior to joining Omeros, Mr. Cancelmo was a principal and shareholder at Garvey Schubert Barer, P.C., where he represented clients in the life sciences and other technology industries in mergers, acquisitions, strategic alliances, public and private securities offerings, and a range of other corporate, commercial and financial transactions. He served as chair of the firm’s business practice group from 2016 until his departure in December 2018. Mr. Cancelmo previously practiced corporate and transactional law at Davies, Ward, Philips and Vineberg LLP, in New York, and Choate, Hall & Stewart LLP, in Boston. Mr. Cancelmo received his J.D. from Boston University and his B.A. from Saint Michael’s College.
Christopher S. Bral, Ph.D. has served as our vice president, nonclinical development since October 2015. From April 2014 to October 2015, Dr. Bral was the executive director, toxicology at Arrowhead Research Corporation, a biopharmaceutical company. From June 2008 to April 2014, Dr. Bral served as director, drug safety evaluation at Vertex Pharmaceuticals, a biotechnology company. Prior to Vertex, Dr. Bral held various pre-clinical drug safety positions of increasing responsibility at Schering-Plough Research Institute including associate director, drug safety evaluation. Dr. Bral received his Ph.D. in biochemistry and biophysics from Texas A&M University and his B.S. in chemistry from John Carroll University. He has been board-certified in toxicology through the American Board of Toxicology since 2000.
Nadia Dac has served as our Chief Commercial Officer since January 2021. Ms. Dac brings nearly three decades of international experience as a strategic commercial leader at large and small biopharmaceutical companies. Prior to joining Omeros, Ms. Dac served as the chief commercial officer at Alder Pharmaceuticals, Inc. (acquired in 2019 by Lundbeck) from April 2019 until June 2020 and as vice president of global specialty commercial development at AbbVie, Inc. from December 2014 to March 2019. She previously served as vice president of marketing at Auxilium Pharmaceuticals, Inc. from May 2013 to September 2014, when the company was acquired by Endo International plc. From 2009 to 2013, Ms. Dac held several roles of increasing responsibility at Novartis AG, including global vice president of neuroscience professional relations prior to her role as vice president of Novartis’ multiple sclerosis franchise, and at Biogen Inc., Johnson & Johnson, and Eli Lilly and Company. She holds a B.S. in Marketing from Rutgers University.
George A. Gaitanaris, M.D., Ph.D. has served as our vice president, science since August 2006 and as our chief scientific officer since January 2012. From August 2003 until our acquisition of nura, inc., in August 2006, Dr. Gaitanaris served as the chief scientific officer of nura, a company that he co-founded, and that developed treatments for central nervous system disorders. From 2000 to 2003, Dr. Gaitanaris served as president and chief scientific officer of Primal, Inc., a biotechnology company that was acquired by nura in 2003. Prior to co-founding Primal, Dr. Gaitanaris served as staff scientist at the National Cancer Institute. Dr. Gaitanaris received his Ph.D. in cellular, molecular and biophysical studies and his M.Ph. and M.A. from Columbia University and his M.D. from the Aristotelian University of Greece.
26
Bruce Meiklejohn, Ph.D. has served as our vice president, chemistry, manufacturing and controls (“CMC”) since October 2019. Prior to joining Omeros in this role, Dr. Meiklejohn was an expert CMC consultant for several biotechnology companies, including Omeros. His consulting work followed a career of over 27 years at Eli Lilly and Company, where he held a number of CMC leadership roles including head of Lilly’s biopharmaceutical product development division and senior research fellow in regulatory affairs CMC. While at Lilly, Dr. Meiklejohn led or played a key role in CMC activities for a number of multibillion-dollar drugs, including Trulicity®, Cialis®, Alimta®, Forteo®, and Cymbalta®. Dr. Meiklejohn earned his Ph.D. in analytical chemistry and his B.S. in biology and chemistry at Colorado State University.
Catherine A. Melfi, Ph.D. has served as our vice president, regulatory affairs and quality systems since October 2012 and has served as our chief regulatory officer since April 2016. Dr. Melfi previously served from January 1996 to September 2012 at Eli Lilly and Company, where she held technical and leadership roles of increasing scope and responsibility, including as senior director and scientific director in global health outcomes and regulatory affairs, respectively. Prior to joining Eli Lilly, Dr. Melfi held various faculty and research positions at Indiana University, including appointments in its Economics Department, in the School of Public and Environmental Affairs, and in the Indiana University School of Medicine. Dr. Melfi received her Ph.D. in Economics from the University of North Carolina - Chapel Hill and B.S. in Economics from John Carroll University.
Tina Quinton, J.D., M.S. has served as our vice president, patents, since June 2019 and previously served as our deputy general counsel, patents from August 2017 to June 2019 and as associate general counsel, patents from 2012 to 2017. Prior to joining Omeros, Ms. Quinton was a partner with the firm Christensen O'Connor Johnson & Kindness, PLLC, where she represented clients in the biotechnology and medical sciences industries in all aspects of worldwide patent procurement and enforcement. Before Christensen O'Connor Johnson & Kindness, Ms. Quinton was a research scientist at several biotechnology companies and centers, including ZymoGenetics, Targeted Genetics Corporation and Fred Hutchinson Cancer Research Center. Ms. Quinton received her J.D. and her M.S. in Molecular and Cellular Biology from the University of Washington and her B.S. from Gordon College.
J. Steven Whitaker, M.D., J.D. has served as our vice president, clinical development since joining Omeros in 2010, and served as our chief medical officer from March 2010 to August 2018 and since November 2019. From May 2008 to March 2010, Dr. Whitaker served as the chief medical officer, vice president of clinical development at Allon Therapeutics, Inc., a biotechnology company focused on developing drugs for neurodegenerative diseases. From August 2007 to May 2008, he served as a medical consultant to Accelerator Corporation, a biotechnology-company investor and incubator. From May 1994 to May 2007, Dr. Whitaker served at ICOS Corporation, which was acquired by Eli Lilly and Company in 2007. At ICOS, he held roles of increasing responsibility in clinical research and medical affairs, most recently as divisional vice president, clinical research as well as medical director of the Cialis® global product team. Dr. Whitaker received his M.D. from the Indiana University School of Medicine, his J.D. from the University of Washington and his B.S. from Butler University.
Peter W. Williams has served as our vice president, human resources since June 2020. Prior to joining Omeros, Mr. Williams served as the senior vice president of human resources at Redbox Automated Retail, LLC from 2016 to 2019, where he led human resources and internal communications functions. From 2013 to 2016, Mr. Williams served as the vice president, HR operations at Outerwall Inc. (Coinstar) and before that he held human resources leadership roles at Coinstar from 2009 to 2013. Prior to 2009, Mr. Williams held human resources leadership roles at various technology and consumer focused companies, including Washington Mutual, Inc., Sterling Commerce, Inc., Expedia, Inc., and Verio, Inc. Mr. Williams received a B.A. in Business Administration and a B.A. in English from the University of Washington.
Corporate Information
We were incorporated in 1994 as a Washington corporation. Our principal executive offices are located at 201 Elliott Avenue West, Seattle, Washington, 98119, and our telephone number is (206) 676-5000. Our website address is www.omeros.com. We make available, free of charge through our investor relations website at investor.omeros.com, our annual report on Form 10-K, our quarterly reports on Form 10-Q, our current reports on Form 8-K and amendments to those reports filed or furnished pursuant to Section 13(a) or 15(d) of the Exchange Act, including exhibits to those
27
reports, as soon as reasonably practicable after we electronically file such material with, or furnish it to, the SEC. Our websites and the information contained therein or incorporated therein are not intended to be incorporated into this Annual Report on Form 10-K. The SEC maintains a website that contains reports, proxy and information statements, and other information regarding reports that we file or furnish electronically with them at www.sec.gov.
SUMMARY RISK FACTORS
The risk factors described below are a summary of the principal risk factors associated with an investment in our company. These are not the only risks we face. You should carefully consider the risk factors discussed in this summary, as well as the risk factors described in Item 1A. of this Annual Report on Form 10-K.
Risks related to our drug candidates, programs and operations include, but are not limited to, the following:
| ● | the magnitude and duration of future royalties paid to us based on net sales by Rayner of OMIDRIA, which are heavily dependent on the continuation of separate payment for OMIDRIA under Medicare Part B, as well as Rayner’s ability to successfully market and sell OMIDRIA in the U.S. and Europe; |
| ● | failure to obtain and maintain regulatory approval for marketing of future commercial products in the U.S. or in foreign jurisdictions; |
| ● | the success of our clinical trials evaluating narsoplimab for treatment of COVID-19 and, even if successful, our ability to manufacture narsoplimab in quantities adequate |
| ● | the impact of the COVID-19 pandemic on our business, operations and financial results as well as significant uncertainty around the evaluation of narsoplimab as a potential treatment for critically ill COVID-19 patients; |
| ● | lack of adequate coverage or reimbursement from government and/or private payers for any drug candidates that we commercialize in the future; |
| ● | unpredictability of our operating results; |
| ● | our ability to raise capital when needed; |
| ● | any failure to comply with current or future government regulations; |
| ● | lack of internal manufacturing capacity and reliance on third parties to manufacture, finish, store and ship supplies of our drug candidates for clinical and, after approval, commercial use; |
| ● | ability to acquire ingredients, excipients, test kits and other materials to manufacture our drug candidates on commercially reasonable terms; |
| ● | delays, suspensions or terminations of our clinical trials or clinical protocols; |
| ● | failure to capitalize on drug candidates or indications; |
| ● | whether our drug candidates will successfully complete clinical development or be suitable for successful commercialization or generation of revenue; |
| ● | substantial costs as a result of commercial disputes, claims, litigation or other legal proceedings; |
| ● | ability to protect our intellectual property and proprietary technologies; |
28
| ● | our indebtedness and liabilities, which could limit the cash flow available for our operations; |
| ● | competition with companies with more resources and experience; |
| ● | reliance on members of our management team and our ability to recruit and retain key personnel; and |
| ● | reliance on third parties to conduct portions of our preclinical research and clinical trials. |
General risks related to our business include the following:
| ● | cyber-attacks or failures in telecommunications or other information technology systems; |
| ● | volatility of our stock price; |
| ● | dilution to our existing shareholders if we issue additional shares of our common stock or other securities that may be convertible into, or exercisable for, our common stock; and |
| ● | the impact of anti-takeover provisions in our charter documents and under Washington law on potential acquisitions of our company. |
ITEM 1A. RISK FACTORS
The risks and uncertainties described below may have a material adverse effect on our business, prospects, financial condition or operating results. In addition, we may be adversely affected by risks that we currently deem immaterial or by other risks that are not currently known to us. You should carefully consider these risks before making an investment decision. The trading price of our common stock could decline due to any of these risks and you may lose all or part of your investment. In assessing the risks described below, you should also refer to the other information contained in this Annual Report on Form 10-K.
Risks Related to Our Products, Programs and Operations
Our ability to achieve profitability is highly dependent on the royalty income we could expect to receive from the sales of OMIDRIA, and to the extent OMIDRIA is not successful, our business, financial condition and results of operations may be materially adversely affected and the price of our common stock may decline.
We are entitled to receive royalty income at 50% of net product sales of OMIDRIA by Rayner until the earlier of January 1, 2025 or the date that separate payment for OMIDRIA under Medicare Part B is secured for a continuous period of at least four years. For the three and 12 months ended December 31, 2021, we reclassified to discontinued operations net sales of OMIDRIA of $30.8 million and $110.7 million, respectively. Royalty income from Rayner for sales of OMIDRIA may not be sufficient to fund our current operations fully and we cannot provide assurance that royalty income from Rayner will be sufficient to fund our operations fully in the future. In the event that royalties from Rayner are insufficient now or in the future, we will need to generate substantially more royalty or milestone income from Rayner or generate other revenue such as through sales of future approved products to achieve and sustain profitability. Sales-based royalty income may be affected by any number of factors, including:
| ● | whether CMS will maintain its current payment policies, which can be revised through annual rulemaking and associated comment periods, and will continue to pay separately under Medicare Part B for non-opioid pain management drugs like OMIDRIA when used during surgery in the ASC setting, as the U.S. base royalty rate would be reduced to 10% during any specific period in which OMIDRIA is no longer eligible for separate payment under Medicare Part B and procedures utilizing OMIDRIA would likely decline significantly, further reducing royalty income; |
29
| ● | whether and when separate payment for OMIDRIA may be secured for a continuous period of at least four years prior to January 1, 2025; |
| ● | whether, and to what extent, if any, we derive royalties from the sale of OMIDRIA outside the U.S.; |
| ● | pricing, coverage and reimbursement policies of government and private payers such as Medicare, Medicaid, the U.S. Department of Veterans Affairs, group purchasing organizations, insurance companies, health maintenance organizations and other plan administrators; |
| ● | a lack of acceptance by physicians, patients and other members of the healthcare community; |
| ● | interruptions in supply of OMIDRIA from our contract manufacturing partners; |
| ● | the availability, relative price and efficacy of the product as compared to alternative treatment options or branded, compounded or generic competing products; |
| ● | an unknown safety risk; |
| ● | changed or increased regulatory restrictions in the U.S., EU and/or other foreign territories. |
Failure to obtain and maintain regulatory approval in the U.S. or in foreign jurisdictions would prevent us from commercializing and marketing our drug candidates.
The regulatory process is subject to substantial agency discretion and risks, including those described herein and elsewhere in these “Risk Factors.” In October 2021, we received a CRL from FDA regarding our BLA for narsoplimab for the treatment of HSCT-TMA. In the CRL, FDA expressed difficulty in estimating the treatment effect of narsoplimab in HSCT-TMA and asserted that additional information will be needed to support regulatory approval. In February 2022, we had a Type A meeting with FDA to discuss the CRL, including each of the review issues that FDA identified as presenting difficulties interpreting the treatment response in the pivotal trial. We are currently awaiting FDA’s response to our rebuttals to each of those review issues. Although we believe that our BLA, as submitted, merits approval and that the data meet or exceed the threshold for substantial evidence of effectiveness, there can be no assurance that the path to approval will not be delayed, and any such path to approval may be costly, require significant time and may not result in approval. Ultimately, we cannot guarantee that FDA will ever approve narsoplimab for the treatment of HSCT-TMA or any other indication.
We also intend to market outside the U.S. any of our drug candidates that are approved in the future. In order to market our products in non-U.S. jurisdictions, we or our partners must obtain separate regulatory approvals and comply with numerous and varying regulatory requirements. The regulatory approval procedure varies among countries and can involve additional testing and data review. The requirements governing marketing authorization, the conduct of clinical trials, pricing and reimbursement vary from country to country. Approval by FDA does not ensure approval by the EMA, and approval by one foreign regulatory authority does not ensure approval by regulatory agencies in other foreign countries or by FDA. The time required to obtain regulatory approval outside the U.S. and EU may differ from that required to obtain FDA or EU approval. The foreign regulatory approval process may include all of the risks associated with obtaining FDA approval discussed in these “Risk Factors” and we may not obtain foreign regulatory approvals on a timely basis, or at all. In addition, even if we were able to obtain regulatory approval for a product in one or more foreign jurisdictions, we may need to complete additional requirements to maintain that approval and our ability to market the product in the applicable jurisdiction.
Clinical trials evaluating narsoplimab for treatment of COVID-19 may be unsuccessful and, even if successful, we may be unable to manufacture narsoplimab in quantities adequate to meet demand.
Narsoplimab has been used to treat approximately 23 critically ill COVID-19 patients under our compassionate use program with highly positive results. However, we cannot provide assurance that the results observed in the
30
compassionate use program will be observed in any future study of narsoplimab for this indication, including the I-SPY COVID-19 trial, or that we will receive regulatory authorization or approval for narsoplimab in the treatment of COVID-19 patients.
Narsoplimab or any other therapeutic candidate that we may develop to treat COVID-19 will be subject to risks in addition to those normally associated with pharmaceutical research, development, and commercialization, such as higher risk of technical failure, lower and transient opportunities for revenue, higher manufacturing costs, product safety or efficacy risks related to an expedited research and development timeline, and novel liability theories. Results from the I-SPY COVID-19 trial may be unfavorable or inconclusive or, even if the results are favorable, FDA or other regulatory bodies may require that we conduct a large-scale trial of narsoplimab in COVID-19 patients, in addition to the I-SPY COVID-19 trial to grant any approval or authorization. These risks may affect our ability to develop or commercialize a therapeutic for COVID-19.
Additionally, contract manufacturing capacity and supplies of raw materials necessary for the production of narsoplimab are limited and we may be unable to secure the large-scale manufacturing capacity from third parties necessary to manufacture narsoplimab in sufficient quantities to enable broader availability of narsoplimab for COVID-19 patients. In addition, widespread vaccination and/or the availability of alternative therapies for COVID-19 could lead to the diversion of governmental and other potential sources of funding or other manufacturing assistance away from us and toward COVID-19 vaccines or other therapeutics and/or limit the commercial viability of narsoplimab for the treatment of COVID-19.
The spread of COVID-19 and efforts to reduce its transmission may negatively impact our business, operations and financial results.
The COVID-19 pandemic has significantly affected the global economy and has adversely affected our prior sales of OMIDRIA due to a reduction in the overall volume of cataract surgery and intraocular lens replacement procedures. Although cataract surgeries have resumed to varying degrees in locations throughout the country, if the number of cataract procedures once again becomes meaningfully limited, either by necessity for time-consuming safety protocols, reduction in patient demand, or the imposition of prohibitions on elective surgeries in some localities, then we would expect there to be a corresponding reduction in demand for OMIDRIA and royalty income we may receive in the future.
We may also experience disruptions to our operations due to COVID-19, such as delays or disruptions with respect to manufacturing of clinical or commercial drug substance or drug product and delays in our clinical trials or in the submission or review of regulatory applications. Such delays or disruptions could negatively affect our commercial operations, clinical programs, and research and development. The health of our employees, contractors and other persons on whom we rely may be adversely affected by COVID-19. Although we are taking precautionary measures intended to help minimize the risk of the virus to our employees, these measures may be ineffective or may otherwise adversely affect our productivity. In addition, the conditions created by the pandemic may intensify other risks inherent in our business. Due to the unknown magnitude, duration and outcome of the COVID-19 pandemic, it is not possible to estimate precisely its impact on our business, operations or financial results; however, the impact could be material.
To the extent COVID-19 adversely affects our business, financial condition, and results of operations and global economic conditions more generally, it may also have the effect of heightening many of the other risk factors set forth herein.
If any other product that we develop and commercialize does not receive adequate coverage or reimbursement from governments and/or private payers, or those potential other commercialized products, our prospects for revenue and profitability would suffer.
Our royalty income and potential revenues depend heavily on the pricing, availability and duration of adequate coverage or reimbursement for the use of products that we or our third-party business partners commercialize, including OMIDRIA, from government, private and other third-party payers, both in the U.S. and in other countries.
31
Pass-through reimbursement, which allows for separate payment (i.e., outside the packaged payment rate for the surgical procedure) under Medicare Part B, expired for OMIDRIA on October 1, 2020. In December 2020, CMS confirmed that OMIDRIA qualifies for separate payment when used on Medicare Part B patients in the ASC setting under CMS’ policy for non-opioid pain management surgical drugs. CMS made separate payment for OMIDRIA effective retroactively as of October 1, 2020. CMS’ current non-opioid separate payment policy and, as a result, separate payment for OMIDRIA thereunder, like other CMS policies in the OPPS and ASC systems, can be changed by CMS through its OPPS/ASC annual rulemaking and comment process. We believe that CMS will continue its separate payment policy for non-opioid pain management surgical drugs, which has been in effect since 2019, and that OMIDRIA will continue to be separately reimbursed when used in the ASC setting. However, we can provide no guarantee that CMS will continue its separate payment policy in future years. If the future reimbursement status of OMIDRIA continues to be uncertain, then demand for OMIDRIA from ASCs and hospitals may be reduced substantially and would negatively impact the amount of royalty income we receive from net product sales of OMIDRIA.
There may be significant delays in obtaining coverage or reimbursement for newly approved products, and we may not be able to provide data sufficient to be granted adequate coverage or reimbursement. Even when a payer determines that a product is eligible for reimbursement, coverage may be limited to the uses of a product that are either approved by FDA (or, in other countries, the relevant country’s regulatory agency) and/or appear in a recognized drug compendium, or other conditions may apply. Moreover, eligibility for coverage does not mean that any product will be reimbursed at a rate that allows us to make a profit in all cases or at a rate that covers our costs, including research, development, manufacturing, sales and distribution. Increasingly, government and private third-party payers that reimburse for healthcare services and products are requiring that companies provide them with predetermined discounts from list prices and are challenging the prices charged for medical products, which could adversely impact the pricing of our products. Any reduction in reimbursement from Medicare or other government programs may result in a similar reduction in payments from private payers. Pricing may also be adversely affected by changes in the terms, scope and/or complexity of government pricing requirements. Even if we achieve coverage or reimbursement for a product, the initial rate or method at which the product will be reimbursed could become unfavorable to us at the time reimbursement is initiated or in the future or may be of a limited duration. In addition, obtaining acceptable coverage and reimbursement from one payer does not guarantee that we will obtain similar acceptable coverage or reimbursement from another payer.
In non-U.S. jurisdictions, we must obtain separate reimbursement approvals and comply with related foreign legal and regulatory requirements. In some countries, including those in the EU, our products may be subject to government price controls. Pricing negotiations with governmental authorities can take a considerable amount of time and expenditure of resources after the receipt of marketing approval for a product. We provide no assurances that the price of any product in one or more of these countries or regions will allow us to make a profit or cover our costs, including research, development, manufacturing, sales and distribution, and as a result we may decide to delay, potentially indefinitely, initiating sales in the particular country or region.
If the reimbursement or pricing that we are able to obtain and maintain for any product that we develop and commercialize, is inadequate, is significantly delayed or is subject to overly restrictive conditions, our ability to generate revenue, attain profitability and/or commercialize our drug candidates may be impaired and there could be a material adverse effect on our business, financial condition, results of operations and growth prospects and trading price of our stock could decline.
Our operating results are unpredictable and may fluctuate.
Our operating results are difficult to predict and will likely fluctuate from quarter to quarter and year to year. We believe that our quarterly and annual results of operations may be affected by a variety of factors, including:
| ● | the level and timing of royalty income from Rayner, as well as our drug candidates if and when approved and commercialized; |
| ● | the extent of coverage and reimbursement for OMIDRIA which may impact whether or not we receive significant milestone payments and/or royalties; |
32
| ● | the extent of any payments received from collaboration arrangements and development funding as well as the achievement of development and clinical milestones under collaboration and license agreements that we may enter into from time to time and that may vary significantly from quarter to quarter; and |
| ● | the timing, cost and level of investment in our research and development activities as well as expenditures we will or may incur to acquire or develop additional technologies, drug candidates, or in preparation for potential commercialization of our drug candidates. |
Any of these risk factors, should one or more occur, could cause the trading price of our stock to decline.
We have incurred cumulative operating losses since inception. If we are unable to raise additional capital when needed, our commercial operations may be limited and we may be unable to complete the development and commercialization of our drug candidates or to continue our other preclinical development programs.
Our operations have consumed substantial amounts of cash since our incorporation and, as of December 31, 2021, we had an accumulated deficit of approximately $682.1 million. We expect to continue to spend substantial amounts to:
| ● | initiate and conduct clinical trials and manufacture clinical and registration batches for our drug candidates; |
| ● | continue research and development in our programs; |
| ● | make principal, interest and fee payments as required under our 6.25% Convertible Senior Notes due 2023 (the “2023 Notes”) and 5.25% Convertible Senior Notes due 2026 (the “2026 Notes” and together with the 2023 Notes, the “Convertible Notes”); and |
| ● | commercialize and launch drug candidates for which we may receive regulatory approval. |
We expect to continue to incur additional losses until such time as we generate significant revenue from the sale of other commercial products or partnerships. We are unable to predict the extent of any future losses and cannot provide assurance that we will generate sufficient revenue from commercial products in the future to fund our operations fully. If we are unable to generate sufficient revenue from commercialized products or partnership arrangements, we may never become and remain profitable and will be required to raise additional capital to continue to fund our operations. We cannot be certain that additional capital will be available to us on acceptable terms, if at all, when required. Adverse developments to our financial condition or business, as well as disruptions in the global equity and credit markets, may limit our ability to access capital. If we do not raise additional capital when needed through one or more funding avenues, such as debt or equity financings or corporate partnering, we may have to significantly delay, scale back or discontinue the development or commercialization of one or more of our drug candidates or one or more of our preclinical programs or other research and development initiatives. In addition, we may be required to seek collaborators for one or more of our current or future products at an earlier stage than otherwise would be desirable or on terms that are less favorable than otherwise might be available or to relinquish or license on unfavorable terms our rights to technologies or products that we otherwise would seek to develop or commercialize ourselves. We also may have insufficient funds or otherwise be unable to advance our preclinical programs, such as potential new drug targets developed from our GPCR program, to a point where they can generate revenue through partnerships, collaborations or other arrangements. Any of these actions could limit the amount of revenue we are able to generate and harm our business and prospects.
We are subject to extensive government regulation and the failure to comply with these regulations may have a material adverse effect on our operations and business.
Both before and after approval of any product, we and our suppliers, contract manufacturers and clinical investigators are subject to extensive regulation by governmental authorities in the U.S. and other countries, covering, among other things, testing, manufacturing, quality control, clinical trials, post-marketing studies, reporting, risk management plans, labeling, advertising, promotion, distribution, import and export, governmental pricing, price
33
reporting and rebate requirements. Failure to comply with applicable requirements could result in one or more of the following actions: warning letters; unanticipated expenditures; delays in approval or refusal to approve a drug candidate; product recall or seizure; interruption of manufacturing or clinical trials; operating or marketing restrictions; injunctions; criminal prosecution and civil or criminal penalties including fines and other monetary penalties; adverse publicity; and disruptions to our business. Further, government investigations into potential violations of these laws would require us to expend considerable resources and face adverse publicity and the potential disruption of our business even if we are ultimately found not to have committed a violation.
Obtaining FDA approval of our drug candidates requires substantial time, effort and financial resources and may be subject to both expected and unforeseen delays, and there can be no assurance that any approval will be granted on any of our drug candidates on a timely basis, if at all. Even if we discuss with, and obtain feedback from, FDA regarding our proposed clinical trials, clinical data collection protocols and nonclinical studies before initiating those trials or studies, FDA may decide that the design of our clinical trials or clinical data collection protocols as actually run, or our resulting data, are insufficient for approval of our drug candidates and may require us to run additional preclinical, clinical or other studies or perform additional work related to chemistry, manufacturing and controls. In addition, we, FDA or an independent institutional review board or ethics committee may suspend or terminate human clinical trials at any time on various grounds, including a finding that the patients are or would be exposed to an unacceptable health risk or because of the way in which the investigators on whom we rely carry out the trials. We are subject to extensive government regulation of the testing of our investigational products, including the requirement that we conduct all of our clinical trials in accordance with FDA’s GCP requirements and similar requirements outside of the U.S. If we are unable to comply with these requirements, if we are required to conduct additional trials or to conduct other testing of our drug candidates beyond that which we currently contemplate for regulatory approval, if we are unable to complete our clinical trials or other testing successfully, or if the results of these and other trials or tests fail to demonstrate efficacy or raise safety concerns, we may face substantial additional expenses, be delayed in obtaining marketing approval for our drug candidates or may never obtain marketing approval.
We are also required to comply with extensive governmental regulatory requirements after a product has received marketing authorization. Governing regulatory authorities may require post-marketing studies that may negatively impact the commercial viability of a product. Once on the market, a product may become associated with previously undetected adverse effects and/or may develop manufacturing difficulties. We are required to comply with other post-marketing requirements including current Good Manufacturing Practices, advertising and promotion restrictions, pharmacovigilance requirements including risk management activities, reporting and recordkeeping obligations, and other requirements. As a result of any of these or other problems or failure to comply with our regulatory obligations, a product’s regulatory approval could be withdrawn, which could harm our business and operating results. In addition, we must maintain an effective healthcare compliance program in order to comply with U.S. and other laws applicable to marketed drug products and, in particular, laws (such as the Anti-Kickback Statute, the False Claims Act and the Sunshine Act) applicable when drug products are purchased or reimbursed by a federal or state healthcare program. U.S. laws such as the Foreign Corrupt Practices Act prohibit the offering or payment of bribes or inducements to foreign public officials, including potentially physicians or other medical professionals who are employees of public healthcare entities in jurisdictions outside the U.S. In addition, many countries have their own laws similar to the healthcare compliance laws that exist in the U.S. Implementing and maintaining an effective compliance program requires the expenditure of significant time and resources. If we are found to be in violation of any of these laws, we may be subject to significant penalties, including but not limited to civil or criminal penalties, damages and fines as well as exclusion from government healthcare programs.
We may face difficulties from changes to current regulations as well as future legislation.
Existing regulatory policies may change and additional government regulations may be enacted that could prevent, limit or delay regulatory approval of our drug candidates. We cannot predict the likelihood, nature or extent of government regulation that may arise from future legislation or administrative action, either in the U.S. or abroad. If we are slow or unable to adapt to changes in existing requirements or the adoption of new requirements or policies, or if we are not able to maintain regulatory compliance, we may lose any marketing approval that we may have obtained and we may not achieve or sustain profitability.
34
There is uncertainty with respect to the impact that healthcare reform legislation and the policies of the Biden administration may have on coverage and reimbursement for healthcare items and services covered by plans that are authorized by the Affordable Care Act (the “ACA”). We expect that the ACA, if it remains in effect, as well as other healthcare reform measures that may be adopted in the future, may result in more rigorous coverage criteria and apply downward pressure on the price that we receive for any approved product. Any reduction in reimbursement from Medicare or other government programs may result in a similar reduction in payments from private payers. If the ACA were to be invalidated or repealed, any resulting reduction in the percentage of the U.S. population that has healthcare insurance could reduce the market for our products. The implementation of cost containment measures or other healthcare reforms may prevent us from being able to generate sufficient revenue, attain and/or maintain profitability or commercialize our drug candidates. We cannot be sure whether additional legislative changes will be enacted, or whether FDA regulations, guidance or interpretations will be changed, or what the impact of such changes on OMIDRIA or the marketing approvals of our drug candidates, if any, may be.
We have no internal capacity to manufacture commercial or clinical supplies of our drug candidates and intend to continue to rely solely on third-party manufacturers. If the contract manufacturers that we rely on experience difficulties manufacturing and supplying our drug candidates, or fail FDA or other regulatory inspections, our clinical trials or regulatory submissions may be significantly limited or delayed.
We rely and intend to continue to rely on third-party manufacturers to produce quantities of clinical drug supplies of our drug candidates that are needed for clinical trials and to support NDAs, BLAs, or similar applications to regulatory authorities seeking marketing approval for our drug candidates, as well as to produce inventory of our drug candidates for commercial use in anticipation of marketing approval. We cannot provide any assurance that we will be able to enter into or maintain these types of arrangements on commercially reasonable terms, or at all. If we or one of our manufacturers were to terminate one of these arrangements early, or the manufacturer was unable to supply product quantities sufficient to meet our requirements, we would be required to transfer manufacturing to an approved alternative facility and/or establish additional manufacturing and supply arrangements. We may also need to establish additional or replacement manufacturers, potentially with little or no notice, in the event that one of our manufacturers fails to comply with FDA and/or other pharmaceutical manufacturing regulatory requirements. Even if we are able to establish additional or replacement manufacturers, identifying these sources and entering into definitive supply agreements and obtaining regulatory approvals may require a substantial amount of time and cost and may create a shortage of the product. It can take several years to qualify and validate a new contract manufacturer, and we cannot guarantee that we would be able to complete in a successful and timely manner the appropriate validation processes or obtain the necessary regulatory approvals for one or more additional or replacement manufacturers. Such alternate supply arrangements may not be available on commercially reasonable terms, or at all. Additionally, if we are unable to engage multiple suppliers to manufacture our products, we may have inadequate supply to meet demand for our product.
In addition, narsoplimab is a biologic drug product and any other drug candidate from certain of our programs, including but not limited to MASP-2 and MASP-3, could be a biologic drug product. We do not have the internal capability to produce biologics for use in clinical trials or on a commercial scale. There are only a limited number of manufacturers of biologic drug products and we may be unable to enter into agreements on commercially reasonable terms with a sufficient number of them to meet clinical or commercial demand, if at all. The regulatory requirements for commercial supply are more stringent than for clinical supply and we cannot guarantee that a contract manufacturer producing drug product for clinical trials will be able to complete successfully the appropriate validation processes or obtain the necessary regulatory approvals for marketing approval and commercial supply in a timely manner or at all.
Our contract manufacturers may encounter difficulties with formulation, manufacturing, supply chain and/or release processes that could result in delays in clinical trials and/or regulatory submissions or that could impact adversely the commercialization of our products or drug candidates, as well as in the initiation of enforcement actions by FDA and other regulatory authorities. For example, our manufacturers are required to comply with FDA’s GMP requirements and are subject to periodic inspections by FDA. If our manufacturers are unable to comply with FDA requirements, they may be unable to meet our supply needs. These difficulties also could result in the recall or withdrawal of a product from the market or a failure to have adequate supplies to meet market demand. If the safety or manufacturing quality of any drug candidate supplied by contract manufacturers is compromised due to one or more of those contract manufacturers’ failure to adhere to applicable laws or for other reasons, we may not be able to maintain regulatory approval to run
35
clinical trials or to obtain and maintain regulatory approval for one or more of our drug candidates, which would harm our business and prospects significantly.
Any significant delays in the manufacture and/or supply of clinical or commercial supplies could materially harm our business, financial condition, results of operations and prospects.
Ingredients, excipients, test kits and other materials necessary to manufacture our drug candidates may not be available on commercially reasonable terms, or at all, which may adversely affect the development and commercialization of our drug candidates.
We and our third-party manufacturers must obtain from third-party suppliers the APIs, excipients, and/or other raw materials plus primary and secondary packaging materials necessary for our contract manufacturers to produce our drug candidates for our clinical trials and, to the extent approved or commercialized, for commercial distribution. Although we have entered or intend to enter into agreements with third-party suppliers that will guarantee the availability and timely delivery of APIs, excipients, test kits and materials for our drug candidates, we have not yet entered into agreements for the supply of all such ingredients, excipients, test kits or materials, and we may be unable to secure all such supply agreements or guarantees on commercially reasonable terms, if at all. Even if we were able to secure such agreements or guarantees, our suppliers may be unable or choose not to provide us the ingredients, excipients, test kits or materials in a timely manner or in the quantities required. If Rayner or its third-party manufacturers experience difficulty obtaining the quantities of these ingredients, excipients or materials that are necessary for the manufacture of commercial supplies of OMIDRIA, the amount of royalty income we could expect to receive would be materially and adversely affected. Further, if we or our third-party manufacturers are unable to obtain APIs, excipients, test kits and materials as necessary for our clinical trials or for the manufacture of commercial supplies of our drug candidates, if approved, potential regulatory approval or commercialization would be delayed, which would materially and adversely affect our ability to generate revenue from the sale of our drug candidates.
If our clinical trials or clinical protocols are delayed, suspended or terminated, we may be unable to develop our drug candidates on a timely basis, which would adversely affect our ability to obtain regulatory approvals, increase our development costs and delay or prevent commercialization of approved products.
We cannot predict whether we will encounter problems with any of our completed, ongoing or planned clinical trials or clinical data collection protocols that will cause regulatory agencies, institutional review boards or ethics committees, or us to delay our clinical trials or suspend or delay the analysis of the data from those trials. Clinical trials and clinical data protocols can be delayed for a variety of reasons, including:
| ● | discussions with FDA, the EMA or other foreign authorities regarding the scope or design of our clinical trials or clinical data collection protocols; |
| ● | delays or the inability to obtain required approvals from institutional review boards, ethics committees or other responsible entities at clinical sites selected for participation in our clinical trials; |
| ● | delays in enrolling patients into clinical trials, collecting data from enrolled patients or collecting historical control data for any reason including disease severity, trial or data collection protocol design, study eligibility criteria, patient population size (e.g., for orphan diseases or for some pediatric indications), proximity and/or availability of clinical trial sites for prospective patients, availability of competing therapies and clinical trials, regional differences in diagnosis and treatment, perceived risks and benefits of the product or drug candidate, disruptions due to external events, including an outbreak of pandemic or contagious disease such as the COVID-19 coronavirus, which has slowed enrollment in our clinical trials of narsoplimab in patients with IgA nephropathy; |
| ● | lower than anticipated retention rates of patients in clinical trials; |
| ● | the need to repeat or conduct additional clinical trials as a result of inconclusive or negative results, failure to replicate positive early clinical data in subsequent clinical trials, failure to deliver an efficacious dose of a drug |
36
| candidate, poorly executed testing, a failure of a clinical site to adhere to the clinical protocol or to follow GCPs or other study requirements, an unacceptable study design or other problems; |
| ● | adverse findings in clinical or nonclinical studies related to the safety of our drug candidates in humans; |
| ● | an insufficient supply of drug candidate materials or other materials necessary to conduct our clinical trials; |
| ● | the need to qualify new suppliers of drug candidate materials for FDA and foreign regulatory approval; |
| ● | an unfavorable inspection or review by FDA or other regulatory authority of a clinical trial site or records of any clinical investigation; |
| ● | the occurrence of unacceptable drug-related side effects or adverse events experienced by participants in our clinical trials; |
| ● | the suspension by a regulatory agency of a trial by imposing a clinical hold; or |
| ● | the amendment of clinical trial or data collection protocols to reflect changes in regulatory requirements and guidance or other reasons as well as subsequent re-examination of amendments to clinical trial or data collection protocols by regulatory agencies, institutional review boards or ethics committees. |
In addition, our clinical trial or development programs have been, and in the future may be, suspended or terminated by us, FDA or other regulatory authorities, or institutional review boards or ethics committees due to a number of factors, including:
| ● | failure to conduct the clinical trial in accordance with regulatory requirements or our clinical protocols; |
| ● | inspection of the clinical trial operations or trial sites by FDA or other regulatory authorities resulting in the imposition of a clinical hold; |
| ● | our failure to comply with our regulatory obligations as a sponsor of clinical research, such as adverse event reporting, control of study drug, adequate study monitoring, and other obligations; |
| ● | the failure to remove a clinical hold in a timely manner, if at all; |
| ● | unforeseen safety issues or any determination that a trial presents unacceptable health risks; |
| ● | inability to deliver an efficacious dose of a drug candidate; or |
| ● | lack of adequate funding to continue the clinical trial or development program, including as a result of unforeseen costs due to enrollment delays, requirements to conduct additional trials and studies and/or increased expenses associated with the services of our contract research organizations (“CROs”), or other third parties. |
If the results of our clinical trials are not available when we expect or if we encounter any delay in the analysis of data from our clinical trials, we may be unable to file for regulatory approval or conduct additional clinical trials on the schedule we currently anticipate. Many of the factors that cause, or lead to, a delay in the commencement or completion of clinical trials may also ultimately lead to the denial of regulatory approval of a drug candidate. Any delays in completing our clinical trials could increase our development costs, could slow down our product development and regulatory submission process, could delay our receipt of product revenue and could make it difficult to raise additional capital. In addition, significant clinical trial delays also could allow our competitors to bring products to market before we do and impair our ability to commercialize our future products, potentially harming our business.
37
Because we have a number of drug candidates and development programs, we may expend our limited resources to pursue a particular drug candidate or indication and fail to capitalize on drug candidates or indications for which there is a greater likelihood of obtaining regulatory approval and that may be more profitable, if approved.
We have limited resources and must focus on the drug candidates and clinical and preclinical development programs that we believe are the most promising. As a result, we may forgo or delay the pursuit of opportunities with other drug candidates or other indications that later prove to have greater commercial potential and may not be able to progress development programs as rapidly as otherwise possible. Further, if we do not accurately evaluate the commercial potential or target market for a particular drug candidate, we may relinquish valuable rights to that drug through collaboration, license or other royalty arrangements in cases in which it would have been advantageous for us to retain sole development and commercialization rights.
Our drug candidates may not successfully complete clinical development or be suitable for successful commercialization or generation of revenue through partnerships, and our preclinical programs may not produce drug candidates that are suitable for clinical trials.
We must successfully complete preclinical testing, which may include demonstrating efficacy and the lack of toxicity in established animal models, before commencing clinical trials for any drug candidate. Many pharmaceutical and biological drug candidates do not successfully complete preclinical testing. There can be no assurance that positive results from preclinical studies will be predictive of results obtained from subsequent preclinical studies or clinical trials. Even if preclinical testing is successfully completed, we cannot be certain that any drug candidates that do advance into clinical trials will successfully demonstrate safety and efficacy in clinical trials. Even if we achieve positive results in early clinical trials, they may not be predictive of the results in later trials, and safety and/or efficacy outcomes of early clinical trials may not be consistent with outcomes of subsequent clinical trials. There can be no assurance that we will be able to successfully commercialize our current or future drug candidates or to meet our expectations with respect to revenues or profits from such products.
We may incur substantial costs as a result of commercial disputes, claims, litigation or other legal proceedings relating to our business operations, especially with regard to patent and other intellectual property rights, and such costs or an adverse outcome in such a proceeding may adversely affect our financial condition, results of operations and/or stock price.
Our business involves numerous commercial contractual arrangements, important intellectual property rights, potential product liability, uncertainties with respect to clinical development, manufacture and regulatory approvals and other aspects that create heightened risks of disputes, claims and legal proceedings. These include claims that may be faced in one or more jurisdictions related to the safety of our drug candidates, the development of our drug candidates, our ability to obtain regulatory approval for our drug candidates, our expectations regarding product development and regulatory approval, sales and marketing practices, commercial disputes including with contract manufacturers, competition, environmental matters, employment matters and other matters. These matters could consume significant time and resources, even if we are successful. Many of our competitors and contractual counterparties are significantly larger than we are and, as a result, may be able to sustain the costs of complex litigation more effectively than we can because they have substantially greater resources. In addition, we may pay damage awards or settlements or become subject to equitable remedies that could, individually or in the aggregate, have a material negative effect on our financial condition, results of operations or stock price. Any uncertainties resulting from the initiation and continuation of any litigation also could have a material adverse effect on our ability to raise the capital necessary to continue our operations.
We may initiate or become subject to litigation regarding patents and other intellectual property rights. Patent infringement litigation involves many complex technical and legal issues and its outcome is often difficult to predict and the risk involved in doing so can be substantial. Generic drug manufacturers could seek approval to market a generic version of our products or challenge our intellectual property rights with respect to our drug candidates.
It may not be feasible to detect and undertake patent enforcement action to stop infringing activity by a number of individual entities, each on a small scale, such as compounding pharmacies. Further, our industry has produced a large number of patents and it is not always clear which patents cover various types of products or methods of use. A third
38
party may claim that we or our contract manufacturers are using inventions covered by the third party’s patent rights and may go to court to stop us from engaging in the alleged infringing activity, including making, using or selling our drug candidates. These lawsuits are costly and could affect our results of operations and divert the attention of managerial and technical personnel. There is a risk that a court would decide that we, or our contract manufacturers, are infringing the third party’s patents and would order us or our contractors to stop the activities covered by the patents. In addition, if we or our contract manufacturers are found to have violated a third party’s patent, we or our contract manufacturers could be ordered to pay damages to the other party. We have agreed to or may agree to indemnify our contract manufacturers against certain patent infringement claims and thus may be responsible for any of their costs associated with such claims and actions. If we were sued for patent infringement, we would need to demonstrate that our drug candidates or methods of use either do not infringe the patent claims of the relevant patent or that the patent claims are invalid, and we might be unable to do this. Proving invalidity, in particular, is difficult since it requires clear and convincing evidence to overcome the presumption of validity enjoyed by issued patents.
It is difficult and costly to protect our intellectual property and our proprietary technologies, and we may not be able to ensure their protection.
Our commercial success will depend in part on obtaining and maintaining patent protection and trade secret protection for the use, formulation and structure of our drug candidates, the methods used to manufacture them, the related therapeutic targets and associated methods of treatment as well as on successfully defending these patents against potential third-party challenges. Our ability to protect our drug candidates from unauthorized making, using, selling, offering to sell or importing by third parties is dependent on the extent to which we have rights under valid and enforceable patents that cover these activities.
The patent positions of pharmaceutical, biotechnology and other life sciences companies can be highly uncertain and involve complex legal and factual questions for which important legal principles remain unresolved. Changes in either the patent laws or in interpretations of patent laws in the U.S. and other countries may diminish the value of our intellectual property. Further, the determination that a patent application or patent claim meets all of the requirements for patentability is a subjective determination based on the application of law and jurisprudence. The ultimate determination by the U.S. Patent and Trademark Office or by a court or other trier of fact in the U.S., or corresponding foreign national patent offices or courts, on whether a claim meets all requirements of patentability cannot be assured. Although we have conducted searches for third-party publications, patents and other information that may affect the patentability of claims in our various patent applications and patents, we cannot be certain that all relevant information has been identified. Accordingly, we cannot predict the breadth of claims that may be allowed or enforced in our patents or patent applications, in our licensed patents or patent applications or in third-party patents.
We cannot provide assurances that any of our patent applications will be found to be patentable, including over our own prior art patents, or will issue as patents. Neither can we make assurances as to the scope of any claims that may issue from our pending and future patent applications nor to the outcome of any proceedings by any potential third parties that could challenge the patentability, validity or enforceability of our patents and patent applications in the U.S. or foreign jurisdictions. Any such challenge, if successful, could limit patent protection for our drug candidates and/or materially harm our business.
The degree of future protection for our proprietary rights is uncertain because legal means afford only limited protection and may not adequately protect our rights or permit us to gain or keep our competitive advantage. In addition, to the extent that we are unable to obtain and maintain patent protection for one of our drug candidates or in the event that such patent protection expires or is limited to method of use patent protection, it may no longer be cost-effective to extend our portfolio by pursuing additional development of a product or drug candidate for follow-on indications.
We also may rely on trade secrets to protect our technologies or drug candidates, especially where we do not believe patent protection is appropriate or obtainable. Although we use reasonable efforts to protect our trade secrets, our employees, consultants, contractors, outside scientific collaborators and other advisers may unintentionally or willfully disclose our information to competitors. Enforcing a claim that a third-party entity illegally obtained and is using any of our trade secrets is expensive and time-consuming, and the outcome is unpredictable. In addition, courts outside the U.S.
39
are sometimes less willing to protect trade secrets. Moreover, our competitors may independently develop equivalent knowledge, methods and know-how.
Our indebtedness and liabilities could limit the cash flow available for our operations and expose us to risks that could adversely affect our business, financial condition and results of operations.
As of December 31, 2021, we had $320.0 million total aggregate principal amount of our 2023 Notes and 2026 Notes outstanding, and we had approximately $1.0 million of outstanding finance lease obligations. We may incur additional indebtedness to meet future financing needs. Our existing and future indebtedness could have significant negative consequences for our security holders and our business, results of operations and financial condition by, among other things:
| ● | requiring a substantial portion of our cash flow from operations to service our indebtedness, which will reduce the amount of cash available for other purposes; |
| ● | limiting our ability to obtain additional financing; |
| ● | limiting our flexibility to plan for, or react to, changes in our business; |
| ● | diluting the interests of our existing stockholders as a result of issuing shares of our common stock upon conversion of the Convertible Notes; |
| ● | placing us at a possible competitive disadvantage with competitors that are less leveraged than we are or have better access to capital; and |
| ● | increasing our vulnerability to adverse economic and industry conditions. |
Our ability to make scheduled payments of the principal of, to pay interest on, or to refinance our indebtedness, including the Convertible Notes, depends on our future performance, which is subject to many factors, including, economic, financial, competitive and other circumstances beyond our control. Our business may not generate sufficient funds, and we may otherwise be unable to maintain sufficient cash reserves, to pay amounts due under our indebtedness, including the Convertible Notes, and our cash needs may increase in the future. In addition, future indebtedness that we may incur may contain, financial and other restrictive covenants that limit our ability to operate our business, raise capital or make payments under our other indebtedness. If we fail to comply with these covenants or to make payments under our indebtedness when due, then we would be in default under that indebtedness, which could, in turn, result in that and our other indebtedness becoming immediately payable in full.
Competitors may develop products that are less expensive, safer or more effective, or which may otherwise diminish or eliminate the success of any products that we may commercialize.
We may not achieve commercial success if our competitors, many of whom have significantly more resources and experience than we, market products that are safer, more effective, less expensive or faster to reach the market than any products that we may develop and commercialize. Our competitors also may market a product that proves to be unsafe or ineffective, which may affect the market for future product we are developing, regardless of the safety or efficacy of our product. The failure of any future product that we may market to compete effectively with products marketed by our competitors would impair our ability to generate revenue, which would have a material adverse effect on our future business, our financial condition and our results of operations.
The loss of members of our management team could substantially disrupt our business operations.
Our success depends to a significant degree on the continued individual and collective contributions of our management team. The members of our management team are at-will employees, and we do not maintain any key-person life insurance policies other than on the life of Gregory A. Demopulos, M.D., our president, chief executive
40
officer and chairman of the board of directors. Losing the services of any key member of our management team, whether from death or disability, retirement, competing offers or other causes, without having a readily available and appropriate replacement could delay the execution of our business strategy, cause us to lose a strategic partner, or otherwise materially affect our operations.
We rely on highly skilled personnel and, if we are unable to retain or motivate key personnel or hire qualified personnel, we may not be able to maintain our operations or grow effectively.
Our performance is largely dependent on the talents and efforts of highly skilled individuals, many of whom possess specialized expertise that may be difficult to replace. Our future success depends on our continuing ability to identify, hire, develop, motivate and retain highly skilled personnel for all areas of our organization. If we are unable to hire and train a sufficient number of qualified employees for any reason, we may not be able to implement our current initiatives or grow effectively. We maintain a rigorous, highly selective and time-consuming hiring process. We believe that our approach to hiring has significantly contributed to our success to date. If we do not succeed in attracting qualified personnel and retaining and motivating existing personnel, our existing operations may suffer and we may be unable to grow effectively.
We may encounter difficulties managing our growth, which could delay our business plans or adversely affect our results of operations.
To manage our future growth, we must continue to implement and improve our managerial, operational and financial systems and continue to recruit, train and retain qualified personnel. We may not be able to implement necessary business processes and systems, recruit, train and retain additional qualified personnel and otherwise manage the growth of our enterprise due to factors such as limited financial resources and competition for qualified personnel within local, national and international markets. The expansion of our operations may lead to significant costs and may divert our management and business development resources. Any inability to manage growth could delay the execution of our business plans or disrupt our operations. Additionally, our inability to manage growth effectively could cause our operating costs to grow even faster than we currently are anticipating.
Our credit facility contains restrictive covenants that may limit our operating flexibility.
In August 2019, we entered into a loan and security agreement with Silicon Valley Bank (“SVB”). The credit facility contains restrictive covenants that limit our ability to transfer or dispose of assets, merge with other companies or consummate certain changes of control, acquire other companies, incur additional indebtedness and liens and enter into new businesses. We therefore may not be able to engage in any of the foregoing transactions unless we obtain the consent of the lender or terminate the credit facility, which may limit our operating flexibility. In addition, our credit facility is secured by all of our assets, excluding our intellectual property and development program inventories. While we had no outstanding borrowings under the credit facility and were in compliance with all covenants as of December 31, 2021, there is no guarantee that we will be able to generate sufficient cash flow or revenue to meet these financial covenants or pay the principal and interest on any future borrowings under our facility.
Product liability claims may damage our reputation and, if insurance proves inadequate, these claims may harm our business.
We may be exposed to the risk of product liability claims that is inherent in the biopharmaceutical industry. A product liability claim may damage our reputation by raising questions about our product’s safety and efficacy and could limit our ability to sell one or more products by preventing or interfering with commercialization of our drug candidates. In addition, product liability insurance for the biopharmaceutical industry is generally expensive to the extent it is available at all. There can be no assurance that we will be able to obtain or maintain such insurance on acceptable terms for any product we bring to market. Further, our product liability insurance coverage may not provide coverage for or may be insufficient to reimburse us for any or all expenses or losses we may suffer. A successful claim against us with respect to uninsured liabilities or in excess of insurance coverage could have a material adverse effect on our business, financial condition and results of operations.
41
We rely on third parties to conduct portions of our preclinical research and clinical trials. If these third parties do not perform as contractually required or otherwise expected, or if we fail to adequately supervise or monitor these parties, we may not be able to obtain regulatory approval for or commercialize our drug candidates.
We rely on third parties, such as CROs, medical and research institutions and clinical investigators, to conduct a portion of our preclinical research, assist us in conducting our clinical trials or to conduct third party-sponsored clinical trials of our drug candidates. Nonetheless, we are responsible for confirming that our preclinical research and clinical trials are conducted in accordance with applicable regulations, the relevant trial protocol and within the context of approvals by an institutional review board or ethics committee, and we may not always be successful in ensuring such compliance. Our reliance on these third parties does not relieve us of responsibility for ensuring compliance with FDA and other regulations and standards for conducting, monitoring, recording and reporting the results of preclinical research and clinical trials to assure that data and reported results are credible and accurate and that the trial participants are adequately protected. If these third parties do not successfully carry out their contractual duties or regulatory obligations or meet expected deadlines, if the third parties need to be replaced or if the quality or accuracy of the data they obtain is compromised due to their failure to adhere to our clinical protocols or regulatory requirements or for other reasons, our preclinical and clinical development processes may be extended, delayed, suspended or terminated, and we may not be able to commercialize or obtain regulatory approval for our drug candidates.
We may need to maintain licenses for active ingredients from third parties to develop and commercialize some of our drug candidates, which could increase our development costs and delay our ability to commercialize those drug candidates.
Should we decide to use APIs in any of our drug candidates that are proprietary to one or more third parties, such as our PDE7 program (OMS527), we would need to maintain licenses to those active ingredients from those third parties. If we are unable to continue to access rights to these active ingredients prior to conducting preclinical toxicology studies intended to support clinical trials, we may need to develop alternate drug candidates from these programs by either accessing or developing alternate active ingredients, resulting in increased development costs and delays in commercialization of these drug candidates. If we are unable to maintain continued access rights to the desired active ingredients on commercially reasonable terms or develop suitable alternate active ingredients, or if we do not meet diligence or other obligations under the corresponding licenses, we may not be able to commercialize drug candidates from these programs.
General Risk Factors Related to our Business
Cyber-attacks or other failures in telecommunications or information technology systems could result in information theft, data corruption and significant disruption of our business operations.
We utilize information technology systems and networks to process, transmit and store electronic information in connection with our business activities. As use of digital technologies has increased, cyber incidents, including deliberate attacks and attempts to gain unauthorized access to computer systems and networks, have increased in frequency and sophistication. These threats pose a risk to the security of our systems and networks, the confidentiality and the availability and integrity of our data. There can be no assurance that we will be successful in preventing cyber-attacks or mitigating their effects. Similarly, there can be no assurance that our collaborators, CROs, third-party logistics providers, distributors and other contractors and consultants will be successful in protecting our clinical and other data that is stored on their systems. Any cyber-attack or destruction or loss of data could have a material adverse effect on our business and prospects. In addition, we may suffer reputational harm or face litigation or adverse regulatory action as a result of cyber-attacks or other data security breaches and may incur significant additional expense to implement further data protection measures.
Our stock price has been and may continue to be volatile, and the value of an investment in our common stock may decline.
During the 12-month period ended December 31, 2021, our stock traded as high as $23.53 per share and as low as $5.67 per share. The trading price of our common stock is likely to continue to be highly volatile and could be subject to
42
wide fluctuations in response to numerous factors, many of which are beyond our control. In addition, the stock market has experienced extreme price and volume fluctuations that have often been unrelated or disproportionate to the operating performance of publicly traded companies. Broad market and industry factors may seriously affect the market price of companies’ stock, including ours, regardless of actual operating performance. These fluctuations may be even more pronounced in the trading market for our stock. In addition, in the past, following periods of volatility in the overall market and the market price of a particular company’s securities, securities class action litigation has often been instituted against these companies. This litigation, if instituted against us, could result in substantial costs and a diversion of our management’s attention and resources.
If we issue additional shares of our common stock or other securities that may be convertible into, or exercisable or exchangeable for, our common stock, our existing shareholders would experience further dilution.
To the extent that we raise additional funds in the future by issuing equity securities, our shareholders would experience dilution, which may be significant and could cause the market price of our common stock to decline significantly. In addition, approximately 13.1 million shares of common stock were subject to outstanding options, awards and warrants as of December 31, 2021 and may become eligible for sale in the public market to the extent permitted by the provisions of various vesting agreements. As of December 31, 2021, we also had approximately 6.0 million shares of common stock reserved for future issuance under our employee benefit plans that are not subject to outstanding options. Further, to the extent we issue common stock upon conversion of the Convertible Notes, such conversion would dilute the ownership interests of existing stockholders despite the expected reduction of such dilution as a result of the capped call transactions that we entered into in connection with the original issuances of the Convertible Notes. If the holders of outstanding options or warrants elect to exercise some or all of them, or if the shares subject to our employee benefit plans are issued and become eligible for sale in the public market, or we issue common stock upon conversion of the Convertible Notes, our shareholders would experience dilution and the market price of our common stock could decline.
Anti-takeover provisions in our charter documents and under Washington law could make an acquisition of us, which may be beneficial to our shareholders, difficult and prevent attempts by our shareholders to replace or remove our current management.
Provisions in our articles of incorporation and bylaws and under Washington law may delay or prevent an acquisition of us or a change in our management. These provisions include a classified board of directors, a prohibition on shareholder actions by less than unanimous written consent, restrictions on the ability of shareholders to fill board vacancies and the ability of our board of directors to issue preferred stock without shareholder approval. In addition, because we are incorporated in Washington, we are governed by the provisions of Chapter 23B.19 of the Washington Business Corporation Act, which, among other things, restricts the ability of shareholders owning 10% or more of our outstanding voting stock from merging or combining with us. Although we believe these provisions collectively provide for an opportunity to receive higher bids by requiring potential acquirers to negotiate with our board of directors, they would apply even if an offer may be considered beneficial by some shareholders. In addition, these provisions may frustrate or prevent any attempts by our shareholders to replace or remove our current management by making it difficult for shareholders to replace members of our board of directors, which is responsible for appointing the members of our management.
We have never declared or paid dividends on our capital stock, and we do not anticipate paying dividends in the foreseeable future.
Our business requires significant funding. We currently plan to invest all available funds and future earnings, if any, in the development and growth of our business. Additionally, under the loan and security agreement with SVB, we have agreed not to pay any dividends. Therefore, we currently do not anticipate paying any cash dividends on our common stock in the foreseeable future. As a result, a rise in the market price of our common stock, which is uncertain and unpredictable, will be the sole source of potential gain for shareholders in the foreseeable future, and an investment in our common stock for dividend income should not be relied upon.
43
ITEM 1B. UNRESOLVED STAFF COMMENTS
None.
ITEM 2. PROPERTIES
We lease approximately 119,719 square feet for our principal office and laboratory space in the building located at 201 Elliott Avenue West, Seattle, Washington (“the Omeros Building”), which includes 9,199 square feet of laboratory space that we are subleasing to third parties. The lease term for our space is through November 2027. We also have two options to extend the lease term, each by five years. The annual base rent due under the lease for our principal office and laboratory space is $7.1 million for 2022, $7.3 million for 2023 and $7.4 million for 2024. In addition, we are responsible for paying our proportionate share of the building’s utilities, taxes, insurance and maintenance as well as a property management fee.
We believe that our facilities are sufficient for our anticipated near-term needs.
ITEM 3. LEGAL PROCEEDINGS
From time to time, in the ordinary course of business, we may be involved in various claims, lawsuits and other proceedings. As of the date of filing of this Annual Report on Form 10-K, we were not involved in any material legal proceedings.
ITEM 4. MINE SAFETY DISCLOSURES
Not applicable.
44
PART II
ITEM 5. MARKET FOR REGISTRANT’S COMMON EQUITY, RELATED SHAREHOLDER MATTERS AND ISSUER PURCHASES OF EQUITY SECURITIES
Market Information
Our common stock is traded on The Nasdaq Global Market under the symbol “OMER.”
Holders
As of February 24, 2022, there were approximately 62,726,515 shares of our common stock outstanding, which were held by 85 holders of record.
Dividends
We have never declared or paid any cash dividends on our capital stock. We expect to retain all available funds and future earnings to fund the development and growth of our business and we do not anticipate paying any cash dividends in the foreseeable future.
Recent Sales of Unregistered Securities
We did not sell any equity securities that were not registered under the Securities Act during the fiscal year ended December 31, 2021.
Stock Performance Graph
The following graph compares the cumulative total shareholder return for our common stock (OMER), the Nasdaq Biotechnology Index (NBI) and the Nasdaq U.S. Benchmark TR Index (NQUSBT) for the period beginning December 31, 2016 and ending December 31, 2021. This graph assumes that $100 was invested on December 31, 2016 in our common stock, the Nasdaq Biotechnology Index and the Nasdaq U.S. Benchmark TR Index. It also assumes that
45
any dividends were reinvested. The data shown in the following graph are not necessarily indicative of future stock price performance.
Comparison of 5 Year Cumulative Return
Assumes Initial Investment of $100
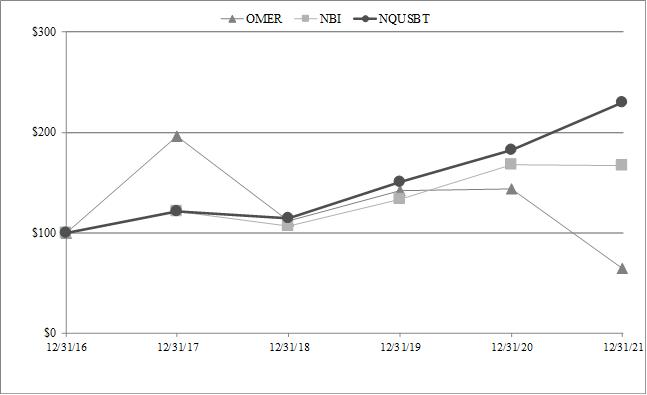
The foregoing information shall not be deemed to be “soliciting material” or to be “filed” for purposes of Section 18 of the Exchange Act or otherwise subject to liability under that Section. In addition, the foregoing information shall not be deemed to be incorporated by reference into any of our filings under the Exchange Act or the Securities Act, except to the extent that we specifically incorporate this information by reference.
ITEM 6. [RESERVED]
ITEM 7. MANAGEMENT’S DISCUSSION AND ANALYSIS OF FINANCIAL CONDITION AND RESULTS OF OPERATIONS
The following discussion and analysis should be read in conjunction with the audited annual consolidated financial statements and the related notes thereto included elsewhere in this Annual Report on Form 10-K. This discussion contains forward-looking statements reflecting our current expectations that involve risks and uncertainties. Actual results may differ materially from those discussed in these forward-looking statements due to a number of factors, including those set forth in the section entitled “Risk Factors” and elsewhere in this Annual Report on Form 10-K. For further information regarding forward-looking statements, please refer to the special note regarding forward-looking statements at the beginning of this Annual Report on Form 10-K. Throughout this discussion, unless the context specifies or implies otherwise, the terms “Company,” “we,” “us” and “our” refer to Omeros Corporation and our wholly owned subsidiaries.
46
Overview
We are a clinical-stage biopharmaceutical company committed to discovering, developing and commercializing small-molecule and protein therapeutics for large-market as well as orphan indications targeting immunologic diseases, including complement-mediated diseases and cancers related to dysfunction of the immune system, as well as addictive and compulsive disorders.
Our drug candidate narsoplimab is the subject of a biologics license application (“BLA”) that, following receipt of a CRL, is pending before the the U.S. Food and Drug Administration (“FDA”) for the treatment of hematopoietic stem cell transplant-associated thrombotic microangiopathy (“HSCT-TMA”). We also have multiple Phase 3 and Phase 2 clinical-stage development programs, which are focused on complement-mediated disorders, including immunoglobulin A (“IgA”) nephropathy, atypical hemolytic uremic syndrome (“aHUS”), and COVID-19. We have successfully completed a Phase 1 clinical trial in healthy subjects and are initiating a Phase 1b clinical trial in PNH patients for our MASP-3 inhibitor OMS906 targeting the alternative pathway of complement. We also have successfully completed a Phase 1 study in our phosphodiesterase 7 (“PDE7”) program focused on addiction. In addition, we have a diverse group of preclinical programs, including GPR174, a novel target in immuno-oncology that modulates a new cancer immunity axis that we discovered. We are also advancing other related cancer therapeutics as well as CAR T-cell and adoptive T-cell therapies. Small-molecule and antibody inhibitors of GPR174 are part of our proprietary G protein-coupled receptor (“GPCR”) platform through which we control 54 GPCR drug targets and their corresponding compounds. We also possess a proprietary-asset-enabled antibody-generating technology.
On December 23, 2021, we closed on an Asset Purchase with Rayner Surgical, Inc. (“Rayner”) for the sale of our commercial product OMIDRIA and certain related assets including inventory and prepaid expenses (the “Transaction”). Rayner paid us $126.0 million in cash at closing, and we retained all outstanding accounts receivable as of the closing date. We will receive a royalty on world-wide sales of OMIDRIA and potentially a $200.0 million milestone payment if separate payment for OMIDRIA is secured in the U.S. for a continuous period of at least four years before January 1, 2025.
As a result of the OMIDRIA divestiture, the results of OMIDRIA operations have been reclassified to net income from discontinued operations, net of tax in our consolidated statements of operations and comprehensive loss and excluded from continuing operations for all periods presented (See Net Income from Discontinued Operations, Net of Tax below for additional information).
As of December 31, 2021, we had $157.3 million in cash and cash equivalents and short-term investments available for general corporate use and $38.2 million in accounts receivable, which we expect to collect in full by March 31, 2022.
Results of Operations
Research and Development Expenses
Our research and development expenses can be divided into three categories: direct external expenses, which include clinical research and development and preclinical research and development activities; internal, overhead and other expenses; and stock-based compensation expense. Direct external expenses consist primarily of expenses incurred pursuant to agreements with third-party manufacturing organizations prior to receiving regulatory approval for a drug candidate, contract research organizations (“CROs”), clinical trial sites, collaborators, and licensors and consultants. Costs are reported in preclinical research and development until the program enters the clinic. Internal, overhead and other expenses consist of personnel costs, overhead costs such as rent, utilities and depreciation and other miscellaneous costs. The discontinued operations of OMIDRIA relates to the costs of drug manufacturing stability and quality control testing and costs of employees and consultants. The following table illustrates our expenses associated with these activities:
47
Year Ended | ||||||||
Year Ended December 31, | ||||||||
2021 | 2020 | 2019 | ||||||
(In thousands) | ||||||||
Continuing research and development expenses: | ||||||||
Direct external expenses: | ||||||||
Clinical research and development: | ||||||||
MASP-2 program - OMS721 (narsoplimab) | $ | 48,806 | $ | 45,020 | $ | 49,804 | ||
MASP-3 program - OMS906 | 7,005 | 7,172 | — | |||||
PDE7 - OMS527 |
| 555 |
| 1,833 |
| 4,066 | ||
Total clinical research and development |
| 56,366 |
| 54,025 |
| 53,870 | ||
Preclinical research and development |
| 15,031 |
| 10,664 |
| 14,291 | ||
Total direct external expenses |
| 71,397 |
| 64,689 |
| 68,161 | ||
Internal overhead and other expenses |
| 40,587 |
| 36,760 |
| 32,155 | ||
Stock-based compensation expense |
| 6,791 |
| 6,163 |
| 6,008 | ||
Total continuing research and development expenses | 118,775 | 107,612 | 106,324 | |||||
Discontinued research and development expense | 3,839 | 3,205 | 3,372 | |||||
Total research and development expenses | $ | 122,614 | $ | 110,817 | $ | 109,696 | ||
Clinical research and development expenses increased $2.3 million between 2021 and 2020 primarily due to increased narsoplimab drug manufacturing costs partially offset by reduced OMS527 toxicology study costs. The change in clinical research and development costs between 2020 and 2019 is primarily due to the migration of OMS906 from preclinical to clinical research and development beginning in the third quarter of 2020 offset by reduced MASP-2 costs.
Preclinical research and development expenses increased $4.4 million in 2021 compared to 2020, primarily due to drug substance, stability and toxicology work on OMS1029 offset by the migration of OMS906 from preclinical to clinical research and development beginning in the third quarter of 2020. The $3.6 million decrease in preclinical research and development expenses in 2020 compared to 2019 was primarily due to the advancement of OMS906 to clinical research and development in the third quarter of 2020.
The increases in internal, overhead and other expenses in all years presented are primarily due to additional employee-related costs and buildout of expanded laboratory facilities to support our research and development activities.
We expect overall continued research and development costs to increase in 2022 as we continue our ongoing Phase 3 clinical programs for narsoplimab and the manufacturing of narsoplimab drug substance to meet our clinical supply needs as well as our commercial requirements should we receive FDA approval for the use of narsoplimab for the treatment of HSCT-TMA. Our accounting policy is to expense all manufacturing costs related to drug candidates until regulatory approval is reasonably assured in either the U.S. or Europe.
At this time, we are unable to estimate with certainty the longer-term costs we will incur in the continued development of our drug candidates due to the inherently unpredictable nature of our preclinical and clinical development activities as well as the potential impact of the COVID-19 pandemic. Clinical development timelines, the probability of success and development costs can differ materially as new data become available and as expectations change. Our future research and development expenses will depend, in part, on the preclinical or clinical success of each drug candidate as well as ongoing assessments of each program’s commercial potential. In addition, we cannot forecast with precision which drug candidates, if any, may be subject to future collaborations, when such arrangements will be secured, if at all, and to what degree such arrangements would affect our development plans and capital requirements.
We are required to expend substantial resources in the development of our drug candidates due to the lengthy process of completing clinical trials and seeking regulatory approval. Any failure or delay in completing clinical trials, or in obtaining regulatory approvals, could delay our generation of product revenue and increase our research and development expenses.
48
Selling, General and Administrative Expenses
Our selling, general and administrative expenses are comprised primarily of salaries, benefits and stock-compensation costs for sales, marketing and other personnel who are not directly engaged in research and development. Costs also include marketing and selling expenses, professional and legal services, general corporate costs and an allocation of our occupancy costs.
Year Ended December 31, | ||||||||
2021 |
| 2020 |
| 2019 | ||||
(In thousands) | ||||||||
Continuing selling, general and administrative expense: | ||||||||
Selling, general and administrative expenses, excluding stock-based compensation expense | $ | 46,688 | $ | 41,692 | $ | 32,755 | ||
Stock-based compensation expense |
| 8,154 |
| 7,614 |
| 6,959 | ||
Total continuing selling, general and administrative expenses | 54,842 | 49,306 | 39,714 | |||||
Discontinued selling, general and administrative expenses | 25,428 | 23,389 | 24,912 | |||||
Total selling, general and administrative expenses | $ | 80,270 | $ | 72,695 | $ | 64,626 | ||
The increase in continuing selling, general and administrative expenses, excluding stock-based compensation, during both years ended December 31, 2021 and 2020 was primarily due to increased pre-commercialization activities for narsoplimab for the treatment of HSCT-TMA.
Our continuing selling, general and administrative expenses for 2022 are highly dependent on the approval of narsoplimab as we have not yet hired the narsoplimab field sales force or initiated various commercial launch activities. If narsoplimab is approved in 2022, our continuing selling, general and administrative expenses will increase as we hire the field sales team and initiate commercial launch activities. If narsoplimab is not approved, our continuing selling, general and administrative expenses are expected to be less than in 2021.
Interest Expense
Year Ended December 31, | ||||||||
2021 |
| 2020 |
| 2019 | ||||
(In thousands) | ||||||||
Interest expense | $ | 19,669 | $ | 26,751 | $ | 22,657 | ||
Interest expense is primarily comprised of contractual interest and amortization of debt issuance and debt discount related to our 6.25% Convertible Senior Notes (the “2023 Notes”) and 5.25% Convertible Senior Notes (the “2026 Notes”) as well as interest on our finance leases. Interest expense decreased $7.1 million compared to the prior year due to the January 1, 2021 adoption of ASU 2020-06, which eliminated the amortization of the non-cash debt discount on the 2023 and 2026 Notes previously allocated to equity. This decrease was partially offset by the increase in interest related to our 2026 Notes, which were issued in August and September 2020. For more information regarding our debt and our unsecured convertible notes (see Part II, Item 8, “Note 9—Unsecured Convertible Senior Notes”).
Loss on Early Extinguishment of Debt
Year Ended December 31, | |||||||||
| 2021 |
| 2020 |
| 2019 | ||||
(In thousands) | |||||||||
Loss on early extinguishment of debt | $ | — | $ | 13,374 | $ | — | |||
In August 2020, we repurchased $115.0 million of the outstanding 2023 Notes. We recorded a $13.4 million loss on early extinguishment of debt related to the unamortized discount and issuance costs related to the repurchase.
49
Other Income
Year Ended December 31, | |||||||||
| 2021 |
| 2020 |
| 2019 | ||||
(In thousands) | |||||||||
Other income | $ | 1,740 | $ | 654 | $ | 1,553 | |||
Other income principally includes sublease rental income and interest earned on our cash and investments. The variations between years is primarily due to $0.8 million of expenses incurred in 2020 in connection with terminating the portion of the capped call related to the 2023 Notes that we repurchased.
Income Tax Benefit
Year Ended December 31, | ||||||||
2021 |
| 2020 |
| 2019 | ||||
(In thousands) | ||||||||
Income tax benefit | $ | — | $ | 23,256 | $ | 19,774 | ||
The income tax benefit in 2020 relates to the issuance of the 2026 Notes respectively (see Part II, Item 8, “Note 14—Income Taxes”).
In December 2019, the Financial Accounting Standards Board issued ASU 2019-12, Income Taxes (Topic 740), which is intended to simplify various aspects of the income tax accounting guidance. ASU 2019-12 eliminates the exception to the incremental approach of intra-period tax allocation when there is a loss from continuing operations and income or gain from other items. As the Company prospectively adopted ASU 2019-12 January 1, 2021, we did not apply any intraperiod allocation rules to 2021. However, we reclassified the tax benefit of income from discontinued operations in prior periods to offset losses from continuing operations.
During 2020, we recorded an income tax benefit of $23.3 million from continuing operations comprising $12.0 million related to the issuance of our 2026 and 2023 Notes, and an additional $11.2 million income tax benefit related to the sale of OMIDRIA assets to Rayner into income from continuing operations. During 2019, we recorded $19.7 million of income tax benefit into continuing operations related to OMIDRIA assets sold to Rayner.
Net Income from Discontinued Operations, Net of Tax
On December 23, 2021, we sold our commercial drug, OMIDRIA, to Rayner. As a result of the OMIDRIA divestiture, the results of OMIDRIA operations have been reclassified to discontinued operations in our consolidated statements of operations and comprehensive loss and excluded from continuing operations for all periods presented. Net income from discontinued operations, net of tax is as follows:
Year Ended December 31, | ||||||||
2021 |
| 2020 |
| 2019 | ||||
(In thousands) | ||||||||
Net income from discontinued operations, net of tax | $ | 385,781 | $ | 35,072 | $ | 62,882 | ||
50
Net income from OMIDRIA operations and the gain recognized on disposition of the asset is shown below:
Year Ended December 31, | |||||||||
| 2021 |
| 2020 |
| 2019 | ||||
(In thousands) | |||||||||
Product sales, net | $ | 110,735 | $ | 73,813 | $ | 111,805 | |||
Royalty income | 1,035 | — | — | ||||||
OMIDRIA income | 111,770 | 73,813 | 111,805 | ||||||
Costs and expenses: |
|
|
|
|
| ||||
Cost of product sales |
| 1,364 |
| 902 |
| 865 | |||
Research and development |
| 3,839 |
| 3,205 |
| 3,372 | |||
Selling, general and administrative |
| 25,428 |
| 23,389 |
| 24,912 | |||
Total costs and expenses |
| 30,631 |
| 27,496 |
| 29,149 | |||
Income before income tax expense |
| 81,139 | 46,317 | 82,656 | |||||
Income tax expense |
| (1,006) |
| (11,245) |
| (19,774) | |||
Net income from discontinued operations, net of tax | 80,133 | 35,072 | 62,882 | ||||||
Gain on sale of OMIDRIA, net | 305,648 | — | — | ||||||
Net income from discontinued operations, net of tax | $ | 385,781 | $ | 35,072 | $ | 62,882 | |||
Product Sales, Net and Royalty Income
The fluctuation in 2020 product sales, net, reclassed to discontinued operations, was due to COVID-19-related reductions in the number of elective cataract procedures from mid-March 2020 through late June 2020. Additionally OMIDRIA pass-through reimbursement under Medicare Part B expired on October 1, 2020 and OMIDRIA revenues were significantly reduced. In December 2020, CMS confirmed that OMIDRIA qualifies for separate payment when used in the ASC setting, and sales normalized during the first half of 2021.
After the sale of OMIDRIA to Rayner, we receive royalty payments of 50% of U.S. domestic net sales. We will continue to earn royalties at this rate until the earlier of January 1, 2025 or when separate payment for OMIDRIA is secured in the U.S. for a continuous period of at least four years. Should separate payment be achieved during this time, the Company would receive a $200.0-million milestone payment from Rayner. Upon the earlier of qualifying for the milestone payment or January 1, 2025, the royalty rate will be reduced to 30% (the “U.S. base royalty rate”) until the expiration or termination of the last issued and unexpired U.S. patent. The U.S. base royalty rate is reduced to 10% upon the occurrence of certain events such as OMIDRIA no longer being eligible for separate payment. We will also receive a royalty of 15% on OMIDRIA net sales outside the U.S. on a country-by-country basis until the expiration or termination of the last issued and unexpired OMIDRIA patent in such country.
OMIDRIA sales have historically been highly dependent on separate payment under Medicare Part B. Given that OMIDRIA reimbursement might be dependent on CMS’ annual renewals and policy, we would likely experience
51
significant fluctuations in period-over-period OMIDRIA royalty earnings should CMS change its non-opioid separate payment policy, which likely would effect CMS’ reimbursement of OMIDRIA.
Deductions to OMIDRIA sales consist of chargebacks, rebates, distribution fees and product return allowances (see Part II, Item 8, “Note 2—Significant Accounting Policies”). The overall percentage deductions to OMIDRIA sales were as follows:
Year Ended December 31, |
| ||||||||
2021 | 2020 | 2019 | |||||||
Deductions percentage to OMIDRIA sales |
| 29.9 | % |
| 31.2 | % |
| 27.7 | % |
The gain on the sale of OMIDRIA included in discontinued operations for the year ended December 31, 2021 is as follows:
(In thousands) | |||
Cash proceeds | $ | 125,993 | |
OMIDRIA contract royalty asset | 184,570 | ||
Gain on sale of OMIDRIA, gross | 310,563 | ||
Transaction and closing costs | (1,972) | ||
Restricted Stock Units ("RSUs") granted to transferred employees | (1,419) | ||
Prepaid assets and inventory at cost | (1,524) | ||
Gain on sale of OMIDRIA, net | $ | 305,648 | |
OMIDRIA Royalties and OMIDRIA Contract Royalty Assets
Upon the closing of the Transaction, we have rights to receive from Rayner future royalties on OMIDRIA net sales at royalty rates that vary based on geography and certain regulatory contingencies. Therefore, future OMIDRIA royalties are treated as variable consideration. The sale of OMIDRIA qualifies as an asset sale. To measure the OMIDRIA contract royalty asset, we used the expected value approach which is the sum of the discounted probability-weighted royalty payments, net of tax, we would receive using a range of potential outcomes, to the extent that it is probable that a significant reversal in the amount of cumulative income recognized will not occur. The contract royalty asset excludes the achievement of the $200.0-million milestone payment and any foreign royalties to the extent it is probable that a significant reversal in the amount of cumulative income recognized will not occur. Royalties earned will be recorded as a reduction to the OMIDRIA contract royalty asset. The amount recorded in discontinued operations in future periods will reflect interest earned on the outstanding OMIDRIA contract royalty asset and any amounts received different from the expected royalties recorded at closing. The OMIDRIA contract royalty asset will also be re-measured periodically using the expected value approach based on actual results and future expectations Any required adjustment to the OMIDRIA contract royalty asset will be recorded into discontinued operations.
On December 22, 2021, the Company granted and expensed RSUs to employees who accepted offers to work for Rayner as a retention incentive to help drive sales of OMIDRIA. The RSUs vest over a two-year period contingent on continued employment at Rayner.
Financial Condition - Liquidity and Capital Resources
As of December 31, 2021, we had $157.3 million in cash, cash equivalents and short-term investments available for general corporate use held primarily in money-market accounts, as compared to $135.0 million at December 31, 2020. As of December 31, 2021, we also had accounts receivable of $38.2 million. We have historically generated net losses and incurred negative cash flows. With the sale of OMIDRIA to Rayner, we had net income of $194.2 million and negative cash flows from operations of $109.7 million as compared to negative cash flows of $100.1 million in the prior year.
52
We plan to continue to fund our operations with our cash and investments, our outstanding accounts receivable, OMIDRIA royalties and potentially the $200.0 million milestone related to achieving long-term OMIDRIA separate payment. If FDA approval is granted for narsoplimab for HSCT-TMA within the next twelve months, sales of narsoplimab will also provide funds for our operations. In addition, we have a sales agreement to sell shares of our common stock, from time to time, in an “at the market” equity offering facility through which we may offer and sell shares of our common stock having an aggregate amount of up to $150.0 million. Should it be determined to be strategically advantageous, we could pursue debt financings as well as public and private offerings of our equity securities, similar to those we have previously completed, or other strategic transactions, which may include licensing a portion of our existing technology. Should it be necessary to manage our operating expenses, we would reduce our projected cash requirements by delaying clinical trials, reducing selected research and development efforts, or implementing other restructuring activities. We have $95.0 million of 2023 Notes due in November 2023. We plan to fund the repayment of the 2023 Notes through cash from operations, including narsoplimab HSCT-TMA revenues should approval be granted by FDA, the $200.0 million milestone related to OMIDRIA, strategic transactions, sale of stock or through issuance of additional debt.
Cash Flow Data
Year Ended December 31, | |||||||||
| 2021 |
| 2020 |
| 2019 | ||||
(In thousands) | |||||||||
Selected cash flow data | |||||||||
Cash provided by (used in): | |||||||||
Operating activities | $ | (109,722) | $ | (100,086) | $ | (60,073) | |||
Investing activities | $ | 193,710 | $ | (67,031) | $ | (3,401) | |||
Financing activities | $ | 6,319 | $ | 174,534 | $ | 60,697 | |||
Operating Activities. Net cash used in operating activities increased for the year ended December 31, 2021 by $9.6 million compared to the same period in 2020. The change in net income adjusted for non-cash items increased by $12.1 million. In addition, we had a $65.7 million increase in the change in operating receivables due to timing of OMIDRIA Medicare Part B reimbursement and a $34.4 million decrease in the change in accounts payable.
Net cash used in operating activities increased for the year ended December 31, 2020 by $40.0 million compared to the same period in 2019. The difference largely resulted from the $53.6 million increase in our net loss from 2019, a $33.0 million increase in cash used in accounts payable and accrued expense, and a $3.6 million increase in cash used for prepaid and other assets. These increases were partially offset by a $43.7 million increase in cash provided from collections of accounts receivable and an increase in non-cash charges of $5.6 million.
Investing Activities. Net cash provided by investing activities increased $260.7 million during 2021 compared to the same period in 2020. This was driven by the $126.0 million payment made as part of the OMIDRIA asset sale and an increase of $134.7 million in net proceeds from the purchase and sale of investments.
Net cash used in investing activities increased $63.6 million during 2020 compared to the same period in 2019, driven by an increase in purchases of investments of $133.2 million offset by proceeds from sale and maturities of investments of $66.4 million.
Financing Activities. Net cash provided by financing activities during 2021 decreased $168.2 million from the prior year. The decrease was due to receiving cash proceeds of $76.9 million, net, in the prior year, from the issuance of our 2026 Notes, which includes the payments for partial repurchase of our 2023 Notes, payments for debt issuance costs, proceeds from termination of our 2023 capped call, and purchases of capped calls related to our 2026 Notes. In addition, we received net proceeds of $93.7 million from our August 2020 public offering of our common stock.
53
Convertible Notes
For more information regarding the 2023 and 2026 Notes see (Part II, Item 8, “Note 8—Unsecured Convertible Senior Notes”).
Line of Credit
We have a Line of Credit Agreement that is secured by all our assets excluding intellectual property and development program inventories and matures on August 2, 2022. The Line of Credit is based upon maintaining a certain amount of accounts receivables including royalty receivables from Rayner. As of December 31, 2021, we had no outstanding borrowings under the Line of Credit Agreement and we were in compliance with all covenants. For more information regarding the Line of Credit Agreement (see Part II, Item 8, “Note 8—Line of Credit”).
Contractual Obligations and Commitments
Operating Leases
We lease our office and laboratory space in The Omeros Building under a lease agreement with BMR - 201 Elliott Avenue LLC. The initial term of the lease ends in November 2027 and we have two options to extend the lease term, each by five years. We lease office and laboratory equipment under various operating and finance lease agreements with initial terms of five years or less. As of December 31, 2021, the remaining aggregate non-cancelable rent payable under the initial term of the lease, excluding common area maintenance and related operating expenses, is $42.9 million.
Convertible Notes
Refer to “Financial Condition—Liquidity and Capital Resources—Convertible Notes” above.
Goods & Services
We have certain non-cancelable obligations under other agreements for the acquisitions of goods and services associated with the manufacturing of our drug candidates, which contain firm commitments. As of December 31, 2021, our aggregate firm commitments are $32.0 million.
We may be required, in connection with in-licensing or asset acquisition agreements, to make certain royalty and milestone payments and we cannot, at this time, determine when or if the related milestones will be achieved or whether the events triggering the commencement of payment obligations will occur. Therefore, such payments are not included in the table above. For information regarding agreements that include these royalty and milestone payment obligations, see Part II, Item 8, “Note 11—Commitments and Contingencies” to our Consolidated Financial Statements in this Annual Report on Form 10-K.
Critical Accounting Policies and Significant Judgments and Estimates
The preparation of our consolidated financial statements, in conformity with U.S. generally accepted accounting principles (“GAAP”), requires management to make estimates and assumptions that affect the amounts reported in the financial statements and accompanying notes. We base our estimates on historical experience and on various other factors that we believe are reasonable under the circumstances; however, actual results could differ from those estimates. An accounting policy is considered critical if it is important to a company’s financial condition and results of operations and if it requires the exercise of significant judgment and the use of estimates on the part of management in its application. Although we believe that our judgments and estimates are appropriate, actual results may differ materially from our estimates. For a summary of our critical accounting policies, (see Part II, Item 8, “Note 2—Significant Accounting Policies” to our Consolidated Financial Statements in this Annual Report on Form 10-K.
54
We believe the following to be our critical accounting policies because they are both important to the portrayal of our financial condition and results of operations and they require critical judgment by management and estimates about matters that are uncertain:
| ● | revenue recognition; |
| ● | OMIDRIA royalties and contract asset accounting; |
| ● | research and development expenses related to clinical trials; |
| ● | accounting for lease agreements, primarily related to our computation of incremental borrowing rate; |
| ● | accounting for convertible debt issuances, primarily related to fair valuing debt and issuance costs; and |
| ● | stock-based compensation, primarily related to our fair value assumptions. |
If actual results or events differ materially from those contemplated by us in making these estimates, our reported financial condition and results of operations for future periods could be materially affected.
Revenue Recognition
Product Sales, Net: We record revenue from product sales when the product is delivered to our wholesalers which is generally when we satisfy all performance obligations. Product sales are recorded net of wholesaler distribution fees and estimated chargebacks, rebates, returns and purchase-volume discounts. Accruals or allowances are established for these deductions in the same period when revenue is recognized, and actual amounts incurred are offset against the applicable accruals or allowances. We reflect each of these accruals or allowances as either a reduction in the related accounts receivable or as an accrued liability depending on how the amount is expected to be settled.
Chargebacks and Rebates: Provisions for chargebacks are determined utilizing historical and projected payer mix and information regarding sell-through and inventory on-hand received directly from wholesalers. Chargebacks are generally settled within four weeks of recording product sales revenue.
We provide reimbursement support services and financial assistance in the form of a rebate to patients whose commercial insurance is inadequate to cover the full cost of our drug product. We apply an experience ratio based on historical and projected patient claims. This experience ratio is applied to product sales to determine the patient rebate accrual and is reviewed and updated periodically to reflect actual results.
Distribution Fees and Product Return Allowances: We pay our wholesalers a distribution fee for services that they perform for us based on the wholesaler average cost value of their purchases. We record a provision against product sales for these charges at the time of sale to the wholesaler.
We allow for the return of product up to 12 months past its expiration date or for product that is damaged. In estimating product returns, we take into consideration our return experience to date, the remaining shelf-life of product we have previously sold, inventory in the wholesale channel and our expectation that product is typically not held by the health care providers based on the frequency of their reorders.
55
OMIDRIA Royalties and OMIDRIA Contract Royalty Asset
Upon the closing of the Transaction, we have rights to receive future royalties from Rayner on OMIDRIA net sales at royalty rates that vary based on geography and certain regulatory contingencies. Therefore, future OMIDRIA royalties are treated as variable consideration. To measure the OMIDRIA contract royalty asset, we used the expected value approach which is the the discounted sum of probability-weighted royalty payments, net of tax, we would receive using a range of potential outcomes, to the extent that it is probable that a significant reversal in the amount of cumulative income recognized will not occur. Our calculations take the net present value of the sum to arrive at the OMIDRIA contract royalty asset stated on the balance sheet. The contract royalty asset excludes the achievement of the $200.0- million milestone payment and any foreign royalties to the extent it is probable that a significant reversal in the amount of cumulative income recognized will not occur. Royalties earned will be recorded as a reduction to the OMIDRIA contract royalty asset. The amount recorded in discontinued operations in future periods will reflect interest earned on the outstanding OMIDRIA contract royalty asset and any amounts received different from the expected royalties recorded at closing. The OMIDRIA contract royalty asset is subject to changes in net sales of OMIDRIA. A 10% change in net sales results in an $18.4 million change in value of the OMIDRIA contract royalty asset, resulting in a potential contract royalty asset valued within the range of $166.7 million to $203.5 million, all else being equal. Changes in net sales could occur due to various risks such as competitors entering the market, technology change as to how cataracts are treated and loss of separate payment status. In determing the value of the OMIDRIA contract royalty asset, we have considered all these factors. The OMIDRIA contract royalty asset will be re-measured periodically using the expected value approach based on actual results and future expectations. Any required adjustment to the OMIDRIA contract royalty asset will be recorded into discontinued operations.
We receive monthly royalty payments based on Rayner’s OMIDRIA product sales in accordance with the Asset Purchase Agreement. Upon the closing of the Transaction, we determined the expected minimum net present value of future OMIDRIA royalty payments and recognized the amount as a gain on the sale of OMIDRIA in discontinued operations on our income statement and as OMIDRIA contract royalty asset on our balance sheet. To determine the OMIDRIA contract royalty asset, we used the expected value approach which is based on the sum of probability-weighted payments we would receive using a range of potential outcomes using a double digit discount rate and the statutory federal income tax rate. The contract royalty asset excludes any revenue which potentially may be reversed in the event of an over estimation. Therefore, we did not include any expectation of receiving the $200.0-million milestone payment or any foreign royalties as we could not judge the probability of those events with certainty.
Royalties earned will be recorded as a reduction to the OMIDRIA contract royalty asset. The amount recorded through earnings in discontinued operations will reflect the time value of money on the outstanding OMIDRIA contract royalty asset. The OMIDRIA contract royalty asset will be evaluated periodically and adjusted using the expected value approach based on actual results and future expectations. Any required adjustments will be recorded into discontinued operations.
Research and Development Expenses
Research and development costs are comprised primarily of:
| ● | contracted research and manufacturing costs; |
| ● | clinical study costs; |
| ● | costs of personnel, including salaries, benefits and stock compensation; |
| ● | consulting arrangements; |
| ● | depreciation and an allocation of our occupancy costs; and |
| ● | other expenses incurred to sustain our overall research and development programs. |
56
Contracted research and manufacturing costs are primarily incurred in the development and production of our drug substance and drug candidates. Prior to approval, our estimates are based on the timing of services provided. We record accrued expenses equal to our estimated expense in excess of amount invoiced by the suppliers.
Clinical trial expenses are estimated on a cost per patient that varies depending on the clinical trial site. As actual costs become known to us, we adjust our estimates; these changes in estimates may result in understated or overstated expenses at any given point in time.
Right-of-Use Assets and Related Lease Liabilities
We record operating leases on our Consolidated Balance Sheet as right-of-use assets and recognize the related lease liabilities equal to the fair value of the lease payments using our incremental borrowing rate when the implicit rate in the lease agreement is not readily available. We derived our incremental borrowing rate by assessing rates in recent market transactions, as adjusted for security interests and our credit quality. A change in the calculated incremental borrowing rate of 100 basis points would not be material to our consolidated financial statements.
Convertible Debt Issuances
On January 1, 2021, we adopted Accounting Standards Update (“ASU”) 2020-06, Debt—Debt with Conversion Options (Subtopic 470.20 and Derivatives and Hedging—Contracts in Entity’s Own Equity (Subtopic 815-40) on a modified retrospective basis. ASU 2020-06 removes the separate liability and equity accounting for our convertible senior notes. As of January 1, 2021, we account for our convertible senior notes wholly as debt. Prior to January 1, 2021, we accounted for convertible debt that may be settled wholly or partially in cash upon conversion as having both a liability component (debt) and an equity component (conversion option). The cash conversion guidance applies as the embedded conversion features meet the requirements for a derivative scope exception for instruments that are both indexed to an entity’s own stock and classified in stockholders’ equity in the balance sheet. Principal cash proceeds from the instrument are allocated first to the liability component based on the fair value of non-convertible debt using the income and market-based approaches to determine an effective interest rate for present valuing the cash proceeds. For the income-based approach, we use a convertible bond pricing model that includes several assumptions such as volatility and a risk-free rate. For the market-based approach, we observe the price of derivative price instruments purchased in conjunction with our convertible senior note issuances or evaluate issuances of convertible debt securities by other companies with similar credit risk ratings at the time of issuance. The amount of the equity component is then calculated by deducting the fair value of the liability component from the principal amount of the instrument. Issuance costs from the instrument are then allocated to the liability and equity components in the same proportion as the proceeds. The equity component of the cash principal proceeds and the liability component of the issuance costs represent a debt discount. which we amortized in prior years as non-cash interest expense over the term of the notes method. On
Transactions involving contemporaneous exchanges of cash between the same debtor and creditor in connection with the issuance of a new debt obligation and satisfaction of an existing debt obligation by the debtor are evaluated as a modification or an exchange transaction depending on whether the exchange is determined to have substantially different terms. The 2023 Notes repurchase and issuance of the 2026 Notes) were deemed to have substantially different terms due to the significant difference between the value of the conversion option immediately prior to and after the exchange. Therefore, the repurchase of the 2023 Notes was accounted for as a debt extinguishment.
Stock-Based Compensation
Stock-based compensation expense is recognized for all share-based payments made to employees, directors and non-employees based on estimated fair values. The fair value of our stock options is calculated using the Black-Scholes option valuation model, which requires assumptions, including volatility, forfeiture rates and expected option life. We estimate forfeitures for expense recognition based on our historical experience. Groups of employees that have similar historical forfeiture behavior are considered separately. If any of the assumptions used in the Black-Scholes model change significantly, stock-based compensation expense for new awards may differ materially from that recorded for existing awards and stock-based compensation for non-employees will vary as the awards are re-measured over the vesting term.
57
Recent Accounting Pronouncements
Please refer to Part II, Item 8, “Note 2--Significant Accounting Policies” to our Consolidated Financial Statements in this Annual Report in Form 10-K for information regarding recent accounting pronouncements.
ITEM 7A. QUANTITATIVE AND QUALITATIVE DISCLOSURES ABOUT MARKET RISK
Our exposure to market risk is primarily confined to our investment securities and debt. The primary objective of our investment activities is to preserve our capital to fund operations, and we do not enter into financial instruments for trading or speculative purposes. We also seek to maximize income from our investments without assuming significant risk. To achieve our objectives, we maintain a portfolio of investments in high-credit-quality securities. As of December 31, 2021, we had cash, cash equivalents and short-term investments of $157.3 million. In accordance with our investment policy, we invest funds in highly liquid, investment-grade securities. The securities in our investment portfolio are not leveraged and are classified as available-for-sale. We currently do not hedge interest rate exposure. Because of the short-term maturities of our investments, we do not believe that an increase in market rates would have a material negative effect on the realized value of our investment portfolio. We actively monitor changes in interest rates and, with our current portfolio of short-term investments, we are not exposed to potential loss due to changes in interest rates.
58
ITEM 8. FINANCIAL STATEMENTS AND SUPPLEMENTARY DATA
Index to Consolidated Financial Statements
Page | |
Report of Independent Registered Public Accounting Firm (PCAOB ID: | 60 |
62 | |
Consolidated Statements of Operations and Comprehensive Loss | 63 |
64 | |
65 | |
66 |
59
Report of Independent Registered Public Accounting Firm
To the Shareholders and the Board of Directors
Omeros Corporation
Opinion on the Financial Statements
We have audited the accompanying consolidated balance sheets of Omeros Corporation (the Company) as of December 31, 2021 and 2020, the related consolidated statements of operations and comprehensive loss, shareholders' equity (deficit) and cash flows for each of the three years in the period ended December 31, 2021, and the related notes (collectively referred to as the “consolidated financial statements”). In our opinion, the consolidated financial statements present fairly, in all material respects, the financial position of the Company at December 31, 2021 and 2020, and the results of its operations and its cash flows for each of the three years in the period ended December 31, 2021, in conformity with U.S. generally accepted accounting principles.
We also have audited, in accordance with the standards of the Public Company Accounting Oversight Board (United States) (PCAOB), the Company's internal control over financial reporting as of December 31, 2021, based on criteria established in Internal Control-Integrated Framework issued by the Committee of Sponsoring Organizations of the Treadway Commission (2013 framework), and our report dated March 1, 2022 expressed an unqualified opinion thereon.
Adoption of ASU No. 2020-06
As discussed in Note 2 to the consolidated financial statements, the Company changed its method of accounting for convertible instruments in 2021 due to the adoption of ASU No. 2020-06, Debt–Debt with Conversion and Other Options (Subtopic 470-20) and Derivatives and Hedging–Contracts in Entity’s Own Equity (Subtopic 815-40): Accounting for Convertible Instruments and Contracts in an Entity’s Own Equity.
Basis for Opinion
These financial statements are the responsibility of the Company's management. Our responsibility is to express an opinion on the Company’s financial statements based on our audits. We are a public accounting firm registered with the PCAOB and are required to be independent with respect to the Company in accordance with the U.S. federal securities laws and the applicable rules and regulations of the Securities and Exchange Commission and the PCAOB.
We conducted our audits in accordance with the standards of the PCAOB. Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the financial statements are free of material misstatement, whether due to error or fraud. Our audits included performing procedures to assess the risks of material misstatement of the financial statements, whether due to error or fraud, and performing procedures that respond to those risks. Such procedures included examining, on a test basis, evidence regarding the amounts and disclosures in the financial statements. Our audits also included evaluating the accounting principles used and significant estimates made by management, as well as evaluating the overall presentation of the financial statements. We believe that our audits provide a reasonable basis for our opinion.
Critical Audit Matters
The critical audit matters communicated below are matters arising from the current period audit of the financial statements that were communicated or required to be communicated to the audit committee and that: (1) relate to accounts or disclosures that are material to the financial statements and (2) involved our especially challenging, subjective, or complex judgments. The communication of critical audit matters does not alter in any way our opinion on the consolidated financial statements, taken as a whole, and we are not, by communicating the critical audit matters below, providing separate opinions on the critical audit matters or on the accounts or disclosures to which they relate.
60
Revenue Deductions | |
Description of the Matter | As more fully described in Note 2 of the consolidated financial statements, product sales to wholesalers are recorded net of revenue deductions. Certain of these revenue deductions require estimates of inventory at wholesalers and ASCs as well as the application of an experience ratio based on historical and projected discounts and rebate claims. Auditing management’s determination of the revenue deductions is complex and requires judgment due to the level of estimation involved in management’s assumptions related to inventories held by wholesalers and ASCs, and the experience ratio used to estimate unsubmitted claims. |
How We Addressed the Matter in Our Audit | We obtained an understanding, evaluated the design, and tested the operating effectiveness of the Company’s internal controls over management’s process for estimating inventories in channel and the experience ratio. To test the revenue deductions, we performed audit procedures that included, among others, evaluating the significant assumptions and the accuracy and completeness of underlying data used in management’s calculations. We compared the significant assumptions used by management to historical ratios of rebate claims to product sales, and other relevant factors. We also assessed the historical accuracy of management’s estimates by comparing previous estimates to actual activity in subsequent periods. |
OMIDRIA Contract Royalty Asset | |
Description of the Matter | As more fully described in Note 2 of the financial statements, the Company recorded a contract asset in connection with its sale of OMIDRIA to Rayner Surgical, Inc. on December 23, 2021. To measure that contract asset, the Company used the expected value approach, which is the sum of the probability-weighted royalty payments using a range of potential outcomes, to the extent that it is probable that a significant reversal in the amount of cumulative income recognized will not occur.
Auditing management’s forecasts is complex and requires judgment due to the level of estimation uncertainty and the sensitivity of the asset’s value to changes in assumptions. In particular, the value of the OMIDRIA contract royalty asset is sensitive to changes in significant assumptions such as forecasted royalties due from Rayner Surgical, Inc. in various scenarios, the probability-weighting of those scenarios, and the discount rate applied, which are affected by expectations about future market and regulatory conditions. |
How We Addressed the Matter in Our Audit | We obtained an understanding, evaluated the design, and tested the operating effectiveness of the Company’s internal controls over management’s process for measuring the OMIDRIA contract royalty asset. To test the measurement of the OMIDRIA contract royalty asset, we performed audit procedures that included, among others, evaluating (1) the estimated future royalties in various scenarios, (2) management’s relative weighting of those scenarios, and (3) the discount rate applied. We compared estimated future royalties to the Company’s historical revenues and royalty rates in the asset purchase agreement. We evaluated the appropriateness and likelihood of occurrence of the various scenarios included in management’s calculation, given the Company’s experience and industry trends. We involved valuation specialists to assist in our testing of the discount rate and verified the clerical accuracy of the calculation. We also evaluated the Company’s disclosures in the consolidated financial statements related to these matters. |
/s/
We have served as the Company’s auditor since 1998.
March 1, 2022
61
OMEROS CORPORATION
CONSOLIDATED BALANCE SHEETS
(In thousands, except share and per share data)
December 31, | December 31, | |||||
| 2021 |
| 2020 | |||
Assets |
|
| ||||
Current assets: |
|
|
|
| ||
Cash and cash equivalents | $ | | $ | | ||
Short-term investments |
| |
| | ||
OMIDRIA contract royalty asset, short-term | | — | ||||
Receivables, net |
| |
| | ||
Prepaid expense and other assets |
| |
| | ||
Current assets from discontinued operations | — | | ||||
Total current assets |
| |
| | ||
OMIDRIA contract royalty asset | | — | ||||
Property and equipment, net |
| |
| | ||
Right of use assets | | | ||||
Restricted investments |
| |
| | ||
Advanced payments, non-current |
| |
| | ||
Total assets | $ | | $ | | ||
Liabilities and shareholders’ equity (deficit) |
|
|
|
| ||
Current liabilities: |
|
|
|
| ||
Accounts payable | $ | | $ | | ||
Accrued expenses |
| |
| | ||
Current portion of lease liabilities |
| |
| | ||
Total current liabilities |
| |
| | ||
Lease liabilities, non-current |
| |
| | ||
Unsecured convertible senior notes, net |
| |
| | ||
Other accrued liabilities - noncurrent |
| |
| — | ||
Commitments and contingencies (Note 11) |
|
|
|
| ||
Shareholders’ equity (deficit): |
|
|
|
| ||
Preferred stock, par value $ |
|
| ||||
Common stock, par value $ |
| |
| | ||
Additional paid-in capital |
| |
| | ||
Accumulated deficit |
| ( |
| ( | ||
Total shareholders’ equity (deficit) |
| |
| ( | ||
Total liabilities and shareholders’ equity (deficit) | $ | | $ | | ||
See accompanying Notes to Consolidated Financial Statements
62
OMEROS CORPORATION
CONSOLIDATED STATEMENTS OF OPERATIONS AND COMPREHENSIVE LOSS
(In thousands, except share and per share data)
Year Ended December 31, | |||||||||
2021 |
| 2020 |
| 2019 | |||||
Product sales, net | $ | — | $ | — | $ | — | |||
Costs and expenses: |
|
|
|
|
| ||||
Cost of product sales |
| — |
| — |
| — | |||
Research and development |
| |
| |
| | |||
Selling, general and administrative |
| |
| |
| | |||
Total costs and expenses |
| |
| |
| | |||
Loss from continuing operations |
| ( |
| ( |
| ( | |||
Loss on early extinguishment of debt |
| — |
| ( |
| — | |||
Interest expense |
| ( |
| ( |
| ( | |||
Other income |
| |
| |
| | |||
Loss from continuing operations before income tax benefit |
| ( |
| ( |
| ( | |||
Income tax benefit |
| — |
| |
| | |||
Net loss from continuing operations | ( | ( | ( | ||||||
Net income from discontinued operations, net of tax | | | | ||||||
Net income (loss) | $ | | $ | ( | $ | ( | |||
Basic and diluted net income (loss) per share | |||||||||
Net loss from continuing operations | ( | ( | ( | ||||||
Net income from discontinued operations | | | | ||||||
Net income (loss) | | ( | ( | ||||||
Weighted-average shares used to compute basic and diluted net income (loss) per share | | | | ||||||
See accompanying Notes to Consolidated Financial Statements
63
OMEROS CORPORATION
CONSOLIDATED STATEMENTS OF SHAREHOLDERS’ EQUITY (DEFICIT)
(In thousands, except share data)
|
| Additional |
|
| Total | |||||||||
Common Stock | Paid-in | Accumulated | Shareholders’ | |||||||||||
Shares | Amount | Capital | Deficit | Equity/(Deficit) | ||||||||||
Balance at December 31, 2018 |
| |
| $ | |
| $ | |
| $ | ( |
| $ | ( |
Issuance of common stock in direct offering, net of offering costs | | | | — | | |||||||||
Issuance of common stock upon exercise of stock options |
| |
| |
| |
| — |
| | ||||
Stock-based compensation |
| — |
| — |
| |
| — |
| | ||||
Net loss |
| — |
| — |
| — |
| ( |
| ( | ||||
Balance at December 31, 2019 |
| |
| |
| |
| ( |
| ( | ||||
Issuance of common stock in direct offering, net of offering costs |
| | | | — | | ||||||||
Issuance of common stock upon exercise of stock options |
| | | | — | | ||||||||
Issuance of common stock upon grant of restricted stock awards | | — | | — | | |||||||||
Stock-based compensation |
| — |
| — |
| |
| — |
| | ||||
Equity component of 2026 Notes, net of issuance costs | — | — | | — | | |||||||||
Purchase of 2026 Capped Calls | — | — | ( | — | ( | |||||||||
Equity component of early extinguishment of 2023 Notes | — | — | ( | — | ( | |||||||||
Termination of the 2023 Capped Call contracts related to debt repurchased | — | — | | — | | |||||||||
Income tax benefit related to issuance of 2026 Notes | — | — | ( | — | ( | |||||||||
Net loss |
| — |
| — |
| — |
| ( |
| ( | ||||
Balance at December 31, 2020 |
| |
| |
| |
| ( |
| ( | ||||
Issuance of common stock upon exercise of stock options and warrants |
| | | | — | | ||||||||
Issuance of common stock upon grant of restricted stock awards | | — | | — | | |||||||||
At the market offering fees | — | — | ( | — | ( | |||||||||
Stock-based compensation |
| — | — | | — | | ||||||||
Cumulative effect of adopting ASU 2020-06 | — | — | ( | ( | ( | |||||||||
Net income |
| — | — | — | | | ||||||||
Balance at December 31, 2021 |
| | $ | | $ | | $ | ( | $ | | ||||
See accompanying Notes to Consolidated Financial Statements
64
OMEROS CORPORATION
CONSOLIDATED STATEMENTS OF CASH FLOWS
(In thousands)
Year Ended December 31, | |||||||||
| 2021 |
| 2020 |
| 2019 | ||||
Operating activities: | |||||||||
Net income (loss) | $ | | $ | ( | $ | ( | |||
Adjustments to reconcile net loss to net cash used in operating activities: |
|
|
|
|
| ||||
Stock-based compensation expense | | | | ||||||
Gain on the sale of OMIDRIA, gross | ( | — | — | ||||||
Non-cash interest expense |
| |
| |
| | |||
Depreciation and amortization |
| |
| |
| | |||
Loss on early extinguishment of debt |
| — |
| |
| — | |||
Deferred income tax |
| — |
| ( |
| — | |||
Fair value settlement upon termination of cap call contract |
| — |
| |
| — | |||
Changes in operating assets and liabilities: |
|
|
|
|
| ||||
Receivables |
| ( |
| |
| ( | |||
Prepaid expenses and other assets |
| |
| ( |
| ( | |||
Accounts payable and other expense |
| |
| ( |
| | |||
Other liabilities non-current |
| |
| — |
| — | |||
Net cash used in operating activities |
| ( |
| ( |
| ( | |||
Investing activities: |
|
|
|
|
|
| |||
Cash proceeds for the sale of OMIDRIA | | — | — | ||||||
Purchases of property and equipment |
| ( |
| ( |
| ( | |||
Purchases of investments |
| ( |
| ( |
| ( | |||
Proceeds from the sale and maturities of investments |
| |
| |
| | |||
Net cash provided by (used in) investing activities |
| |
| ( |
| ( | |||
Financing activities: |
|
|
|
|
|
| |||
Proceeds from issuance of convertible senior notes | — | | — | ||||||
Payments for debt issuance costs | — | ( | — | ||||||
Purchases of capped calls related to convertible senior notes |
| — |
| ( |
| — | |||
Payments for repurchases of convertible senior notes |
| — |
| ( |
| — | |||
Proceeds from termination of capped call contracts |
| — |
| |
| — | |||
Proceeds from issuance of common stock, net | — | | | ||||||
Release in restricted investments |
| — |
| |
| — | |||
Proceeds upon exercise of stock options and warrants |
| |
| |
| | |||
At the market offering costs |
| ( |
| — |
| — | |||
Payments on finance lease obligations |
| ( |
| ( |
| ( | |||
Net cash provided by financing activities |
| |
| |
| | |||
Net increase (decrease) in cash and cash equivalents |
| |
| |
| ( | |||
Cash and cash equivalents at beginning of period |
| |
| |
| | |||
Cash and cash equivalents at end of period | $ | | $ | | $ | | |||
Supplemental cash flow information |
|
|
|
|
|
| |||
Cash paid for interest | $ | | $ | | $ | | |||
Property acquired under finance lease | $ | | $ | | $ | | |||
See accompanying Notes to Consolidated Financial Statements
65
OMEROS CORPORATION
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
Note 1—Organization and Basis of Presentation
General
Omeros Corporation (“Omeros,” the “Company” or “we”) is a clinical-stage biopharmaceutical company committed to discovering, developing and commercializing small-molecule and protein therapeutics for large-market as well as orphan indications targeting immunologic diseases, including complement-mediated diseases and cancers related to dysfunction of the immune system, as well as addictive and compulsive disorders. Our first drug product, OMIDRIA® (phenylephrine and ketorolac intraocular solution) 1% / 0.3%, is marketed in the United States (the “U.S.”) for use during cataract surgery or intraocular lens replacement. We sold OMIDRIA and related business assets on December 23, 2021. See “Sale of OMIDRIA Assets” below for additional information.
Our drug candidate narsoplimab is the subject of a biologics license application (“BLA”) pending before the U.S. Food and Drug Administration (“FDA”) for the treatment of hematopoietic stem cell transplant-associated thrombotic microangiopathy (“HSCT-TMA”). On October 18, 2021, we announced the receipt of a Complete Response Letter (“CRL”) from FDA regarding the BLA. In the CRL, FDA expressed difficulty in estimating the treatment effect of narsoplimab in HSCT-TMA and asserted that additional information will be needed to support regulatory approval. In February 2022, we had a Type A meeting with FDA to discuss the CRL, including each of the review issues that FDA identified as presenting difficulties interpreting the treatment response in the pivotal trial. We are currently awaiting FDA’s response to our rebuttals to each of those review issues. We continue to believe that our BLA, as submitted, merits approval and that the data meet or exceed the threshold for substantial evidence of effectiveness.
We also have multiple late-stage clinical development programs in our pipeline, which are focused on: complement-mediated disorders, including immunoglobulin A (“IgA”) nephropathy, atypical hemolytic uremic syndrome (“aHUS”) and COVID-19.
Sale of OMIDRIA Assets
On December 23, 2021, we closed on an Asset Purchase Agreement (the “Asset Purchase Agreement”) with Rayner Surgical Inc. (“Rayner”) for the sale of our commercial product OMIDRIA and certain related assets including inventory and prepaid expenses (the “Transaction”). Rayner paid us $
As a result of the divestiture, the results of OMIDRIA operations (e.g., revenues and operating costs) have been reclassified to discontinued operations in our consolidated statements of operations and comprehensive loss and excluded from continuing operations for all periods presented (See “Note 3 – Discontinued Operations”).
Basis of Presentation
Our consolidated financial statements include the financial position and results of operations of Omeros and our wholly owned subsidiaries. All inter-company transactions have been eliminated. The accompanying consolidated financial statements have been prepared in accordance with U.S. generally accepted accounting principles (“GAAP”). Certain prior year amounts in the balance sheet, statement of cash flows and the footnotes have been reclassified in the consolidated financial statements to conform to the current year presentation.
Risks and Uncertainties
As of December 31, 2021, we had cash, cash equivalents and short-term investments of $
66
2021 was $
We plan to continue to fund our operations for the next twelve months with our existing cash and investments, our current accounts receivable, and OMIDRIA royalties. There is also the potential for us to receive a $
Management believes the assets on hand along with expected royalties received are adequate to finance our operations at least through March 2, 2023. Accordingly, the accompanying consolidated financial statements have been prepared on a going-concern basis, which contemplates the realization of assets and the satisfaction of liabilities in the normal course of business.
The outbreak of the novel strain of coronavirus that causes COVID-19 and the responses to the global pandemic by various governmental authorities, the medical community and others has had a significant impact on our business. Due to the unknown magnitude, duration, and outcome of the COVID-19 pandemic, it is not possible to estimate precisely the continued impact on our business, operations or financial results; however, the impact has been and could continue to be substantial.
Segments
We operate in
Use of Estimates
The preparation of financial statements in conformity with GAAP requires management to make estimates and assumptions that affect the amounts reported in the financial statements and accompanying notes. Significant items subject to such estimates include revenue recognition, OMIDRIA contract royalty asset valuation, stock-based compensation expense, and accruals for clinical trials and manufacturing of drug product. We base our estimates on historical experience and on various other factors, including the impact of the COVID-19 pandemic, that we believe are reasonable under the circumstances; however, actual results could differ from these estimates.
Note 2—Significant Accounting Policies
Discontinued Operations
We review the presentation of planned or completed business dispositions in the consolidated financial statements based on the available information and events that have occurred. The review consists of evaluating whether the business meets the definition of a component for which the operations and cash flows are clearly distinguishable from the other components of the business and, if so, whether it is anticipated that after the disposal the cash flows of the component would be eliminated from continuing operations and whether the disposition represents a strategic shift that has a major effect on operations and financial results.
Planned or completed business dispositions are presented as discontinued operations when all the criteria described above are met. For those divestitures that qualify as discontinued operations, all comparative periods presented are reclassified in the consolidated balance sheets. Additionally, the results of operations of a discontinued operation are reclassified to income from discontinued operations, net of tax, for all periods presented in the consolidated statements
67
of operations and comprehensive loss. Results of discontinued operations include all revenues and expenses directly derived from such businesses; general corporate overhead is not allocated to discontinued operations. The OMIDRIA asset sale to Rayner qualifies as a discontinued operation and has been presented as such for all reporting periods presented. The Company included information regarding cash flows from discontinued operations (see “Note 3 – Discontinued Operations”).
OMIDRIA Royalties and OMIDRIA Contract Royalty Assets
Upon the closing of the Transaction, we have rights to receive future royalties from Rayner on OMIDRIA net sales at royalty rates that vary based on geography and certain regulatory contingencies. Therefore, future OMIDRIA royalties are treated as variable consideration. The sale of OMIDRIA qualifies as an asset sale. To measure the OMIDRIA contract royalty asset, we used the expected value approach which is the sum of the discounted probability-weighted royalty payments, net of tax, we would receive using a range of potential outcomes, to the extent that it is probable that a significant reversal in the amount of cumulative income recognized will not occur. Accordingly, the contract royalty asset excludes the achievement of the $
Cash and Cash Equivalents, Short-Term Investments and Restricted Investments
Cash and cash equivalents include highly liquid investments with a maturity of three months or less on the date of purchase. Short-term investment securities are classified as available-for-sale and are carried at fair value. Unrealized gains and losses, if any, are reported as a separate component of shareholders’ equity. Amortization, accretion, interest, and dividends, realized gains and losses and declines in value judged to be other-than-temporary are included in other income. The cost of securities sold is based on the specific-identification method. Investments in securities with maturities of less than one year, or those for which management intends to use the investments to fund current operations, are included in current assets. We evaluate whether an investment is other-than-temporarily impaired based on the specific facts and circumstances. Factors that are considered in determining whether an other-than-temporary decline in value has occurred include: the market value of the security in relation to its cost basis; the financial condition of the investee; and the intent and ability to retain the investment for a sufficient period of time to allow for recovery in the market value of the investment. Restricted investments held in money-market funds include security deposits held by our landlord.
As of December 31, 2021 and 2020, all investments are classified as short-term and available-for-sale. Investment income, which is included as a component of other income, consists primarily of interest earned.
Inventory
Inventory is stated at the lower of cost or market determined on a specific identification basis in a manner that approximates the first-in, first-out (“FIFO’) method. Costs include amounts related to third-party manufacturing, transportation, and internal labor and overhead. Capitalization of costs as inventory begins when regulatory approval of the drug candidate is reasonably assured in the U.S. or the European Union (‘EU”). We expense inventory costs related to drug candidates as research and development expenses prior to receiving regulatory approval in the respective territory. Inventory is reduced to net realizable value for excess and obsolete inventories based on forecasted demand. Inventory with an alternative future use is capitalized.
Receivables, Net
Receivables relate primarily to sales of OMIDRIA made to wholesalers prior to the sale to Rayner and include reductions for estimated chargebacks and product returns that are expected to be settled through reductions in
68
receivables. Remaining receivables generally consist of amounts from subleases for space in our facilities. Considering the nature and historic collectability of our receivables, we concluded an allowance for doubtful accounts is not necessary as of December 31, 2021 and 2020.
Property and Equipment, Net
Property and equipment are stated at cost, and depreciation is calculated using the straight-line method over the estimated useful life of the assets, which is generally to
Right-of-Use Assets and Related Lease Liabilities
We record operating leases as right-of-use assets and recognize the related lease liabilities equal to the fair value of the lease payments using our incremental borrowing rate when the implicit rate in the lease agreement is not readily available. We recognize variable lease payments, when incurred. Costs associated with operating lease assets are recognized on a straight-line basis within operating expenses over the term of the lease.
We record finance leases as a component of property and equipment and amortize these assets within operating expenses on a straight-line basis to their residual values over the shorter of the term of the underlying lease or the estimated useful life of the equipment. The interest component of a finance lease is included in interest expense and recognized using the effective interest method over the lease term.
We account for leases with initial terms of 12 months or less as operating expenses on a straight-line basis over the lease term.
Unsecured Convertible Senior Notes
On January 1, 2021, we adopted Accounting Standards Update (“ASU”) 2020-06, Debt—Debt with Conversion Options (Subtopic 470.20 and Derivatives and Hedging—Contracts in Entity’s Own Equity (Subtopic 815-40) on a modified retrospective basis. ASU 2020- removes the separate liability and equity accounting for our convertible senior notes that was required under previous guidance and allows us to account for our convertible senior notes wholly as debt. Upon adoption, we removed the equity component allocated to debt issuance costs increasing unsecured convertible senior notes and shareholders’ equity by $
Transactions involving contemporaneous exchanges of cash between the same debtor and creditor in connection with the issuance of a new debt obligation and satisfaction of an existing debt obligation by the debtor are evaluated as a modification or an exchange transaction depending on whether the exchange is determined to have substantially different terms. The
Impairment of Long-Lived Assets
We assess the impairment of long-lived assets, primarily property and equipment, whenever events or changes in circumstances indicate that the carrying value of an asset may not be recoverable. Recoverability of these assets is measured by comparing the carrying value to future undiscounted cash flows that the asset is expected to generate. If the asset is impaired, the amount of any impairment will be reflected in the results of operations in the period of impairment. We have not recognized any impairment losses for the years ended December 31, 2021, 2020 and 2019.
69
Revenue Recognition
When we enter into a customer contract, we perform the following five steps: (i) identify the contract with a customer; (ii) identify the performance obligations in the contract; (iii) determine the transaction price; (iv) allocate the transaction price to the performance obligations in the contract; and (v) recognize revenue when (or as) we satisfy a performance obligation.
Product Sales, Net
We generally record revenue from product sales when the product is delivered to our wholesalers and title for the product is transferred. Product sales are recorded net of wholesaler distribution fees and estimated chargebacks, rebates, returns and purchase-volume discounts. Accruals or allowances are established for these deductions in the same period when revenue is recognized, and actual amounts incurred are offset against the applicable accruals or allowances. We reflect each of these accruals or allowances as either a reduction in the related accounts receivable or as an accrued liability depending on how the amount is expected to be settled.
Chargebacks and Rebates
Provisions for chargebacks are determined utilizing historical and projected payer mix and information regarding sell-through and inventory on-hand received directly from wholesalers. Chargebacks are generally settled within weeks of recording product sales revenue.
We provide reimbursement support services and financial assistance in the form of a rebate to patients whose commercial insurance is inadequate to cover the full cost of our drug product. We apply an experience ratio based on historical and projected patient claims. This experience ratio is applied to product sales to determine the patient rebate accrual and is being reviewed and updated periodically to reflect actual results.
Distribution Fees and Product Return Allowances
We pay our wholesalers a distribution fee for services that they perform for us based on the wholesaler average cost value of their purchases. We record a provision against product sales for these charges at the time of sale to the wholesaler.
We allow for the return of product up to
Research and Development
Research and development expenses are comprised primarily of contracted research and manufacturing costs prior to approval; costs for personnel, including salaries, benefits and stock compensation; clinical study costs; contracted research; manufacturing costs prior to approval; consulting services; depreciation; materials and supplies; milestones; an allocation of our occupancy costs; and other expenses incurred to sustain our overall research and development programs. Advance payments for goods or services that will be used or rendered for future research and development activities are deferred and then recognized as an expense as the related goods are delivered or the services are performed, or when the goods or services are no longer expected to be provided. All other research and development costs are expensed as incurred.
Selling, General and Administrative
Selling, general and administrative expenses are comprised primarily of salaries, benefits, and stock-compensation costs for sales, marketing, and other personnel not directly engaged in research and development. Additionally, selling, general and administrative expenses include marketing and selling expenses, professional and legal services; patent
70
costs; depreciation, an allocation of our occupancy costs; and other general corporate expenses. Advertising costs, which we consider to be media and marketing materials, are expensed as incurred and were $
Income Taxes
Deferred tax assets and liabilities are recognized for the future tax consequences attributable to differences between the financial statement carrying amounts of existing assets and liabilities and their tax bases. Deferred tax assets and liabilities are measured using enacted tax rates applied to taxable income in the years in which those temporary differences are expected to be recovered or settled. We recognize the effect of income tax positions only if those positions are more likely than not of being sustained upon an examination. A valuation allowance is established when it is more likely than not that the deferred tax assets will not be realized.
Stock-Based Compensation
Stock-based compensation expense is recognized for all share-based payments based on estimated fair values. The fair value of our stock options is calculated using the Black-Scholes option-pricing model which requires judgmental assumptions around volatility, forfeiture rates and expected option term. Compensation expense is recognized over the optionees’ requisite service periods, which is generally the vesting period, using the straight-line method. Forfeiture expense is estimated at the time of grant and revised in subsequent periods if actual forfeitures differ from those estimates.
Accumulated Other Comprehensive Loss
Accumulated other comprehensive loss is comprised of net loss and certain changes in equity that are excluded from net loss. There was no difference between comprehensive loss and net loss for the years ended December 31, 2021, 2020 or 2019.
Financial Instruments and Concentrations of Credit Risk
Cash and cash equivalents, receivables, accounts payable and accrued liabilities, which are recorded at invoiced amount or cost, approximate fair value based on the short-term nature of these financial instruments. The fair value of short-term investments is based on quoted market prices. Financial instruments that potentially subject us to concentrations of credit risk consist primarily of cash and cash equivalents, short-term investments and receivables. Cash and cash equivalents are held by financial institutions and are federally insured up to certain limits. At times, our cash and cash equivalents balance held at a financial institution may exceeds the federally insured limits. To limit the credit risk, we invest our excess cash in high-quality securities such as money market mutual funds, certificates of deposit and commercial paper.
Major Customers
Prior to the sale of OMIDRIA to Rayner, we sold OMIDRIA through a limited number of wholesalers. Each of these wholesalers, together with entities under their common control, accounted for greater than 10% of our total
71
revenues for the years ended December 31, 2021, 2020 and 2019 and greater than 10% of accounts receivable as of December 31, 2021, 2020 and 2019 as noted below.
2021 | 2020 | 2019 | ||||||||||||||||
Percentage | Percentage | Percentage | Percentage | Percentage | Percentage | |||||||||||||
of Total | of Accounts | of Total | of Accounts | of Total | of Accounts | |||||||||||||
Revenue | Receivable | Revenue | Receivable | Revenue | Receivable | |||||||||||||
Distributor A |
| | % | | % | | % | | % | | % | | % | |||||
Distributor B |
| | % | | % | | % | | % | | % | | % | |||||
Distributor C |
| | % | | % | | % | | % | | % | | % | |||||
Distributor D |
| | % | | % | | % | | % | | % | | % | |||||
Note 3—Discontinued Operations
On December 23, 2021, we closed an Asset and Purchase Agreement for the sale of OMIDRIA and certain related assets including inventory and prepaid expenses. We retained the outstanding accounts receivable and all outstanding liabilities related to OMIDRIA as of the closing date.
Upon closing, we received an up-front cash payment of $
The sale of OMIDRIA was recorded as an asset sale and all comparative periods presented are required to be reclassified in the consolidated balance sheets. Additionally, the results of operations for OMIDRIA are reclassified to income from discontinued operations for all periods presented in the consolidated statements of operations and comprehensive loss.
Net income from discontinued operations, net of tax is as follows:
Year Ended December 31, | |||||||||
| 2021 |
| 2020 |
| 2019 | ||||
(In thousands) | |||||||||
Product sales, net | $ | | $ | | $ | | |||
Royalty income | | — | — | ||||||
OMIDRIA income | | | | ||||||
Costs and expenses: |
|
|
|
|
| ||||
Cost of product sales |
| |
| |
| | |||
Research and development |
| |
| |
| | |||
Selling, general and administrative |
| |
| |
| | |||
Total costs and expenses |
| |
| |
| | |||
Income before income tax expense |
| |
| |
| | |||
Income tax expense |
| ( |
| ( |
| ( | |||
Net income from discontinued operations, net of tax | | | | ||||||
Gain on sale of OMIDRIA, net |
| |
| — |
| — | |||
Net income from discontinued operations, net of tax | $ | | $ | | $ | | |||
72
The gain on the sale of OMIDRIA included in discontinued operations for the year ended December 31, 2021 is as follows:
(In thousands) | |||
Cash proceeds | $ | | |
OMIDRIA contract royalty asset | | ||
Gain on sale of OMIDRIA, gross | | ||
Transaction and closing costs | ( | ||
Restricted Stock Units ("RSUs") granted to transferred employees | ( | ||
Prepaid assets and inventory at cost | ( | ||
Gain on sale of OMIDRIA, net | $ | | |
Cash flow from discontinued operations is as follows:
Year Ended December 31, | |||||||||
| 2021 |
| 2020 |
| 2019 | ||||
(In thousands) | |||||||||
Total operating cash flows from discontinued operations | $ | | $ | | $ | ( | |||
Total investing cash flows from discontinued operations | $ | | $ | — | $ | — | |||
Note 4—Net Loss Per Share
Our potentially dilutive securities include potential common shares related to our stock options, warrants, restricted stock units and unsecured convertible senior notes. Diluted earnings per share (“Diluted EPS”) considers the impact of potentially dilutive securities except in periods in which there is a loss because the inclusion of the potential common shares would have an anti-dilutive effect. Diluted EPS excludes the impact of potential common shares related to our stock options in periods in which the option exercise price is greater than the average market price of our common stock for the period.
Potentially dilutive securities excluded from Diluted EPS are as follows:
Year Ended December 31, | |||||
2021 |
| 2020 | 2019 | ||
2023 Notes convertible to common stock (1) | | | | ||
Outstanding options to purchase common stock | |
| | | |
Outstanding restricted stock units | | — | — | ||
Outstanding warrants to purchase common stock | — |
| | | |
Total potentially dilutive shares excluded from loss per share | |
| | | |
| (1) | The 2023 Notes are subject to a capped call arrangement that potentially reduces the dilutive effect as described in “Note 9 — Unsecured Convertible Senior Notes”. Any potential impact of the capped call arrangement is excluded from this table. |
Note 5—Accounts Receivable, Net
Accounts receivable, net consists of the following:
December 31, | December 31, | |||||
| 2021 |
| 2020 | |||
(In thousands) | ||||||
Trade receivables, net | $ | | $ | | ||
Sublease and other receivables |
| |
| | ||
Total accounts receivables, net | $ | | $ | | ||
Trade receivables are shown net of $
73
Note 6—Fair-Value Measurements
As of December 31, 2021 and 2020, all investments were classified as short-term and available-for-sale. Investment income, which was included as a component of other income, consists of interest earned.
On a recurring basis, we measure certain financial assets at fair value. Fair value is defined as the exchange price that would be received for an asset or paid to transfer a liability, an exit price, in the principal or most advantageous market for the asset or liability in an orderly transaction between market participants on the measurement date. The accounting standard establishes a fair value hierarchy that requires an entity to maximize the use of observable inputs, where available. The following summarizes the three levels of inputs required:
Level 1—Observable inputs for identical assets or liabilities, such as quoted prices in active markets;
Level 2—Inputs other than quoted prices in active markets that are either directly or indirectly observable; and
Level 3—Unobservable inputs in which little or no market data exists, therefore they are developed using estimates and assumptions developed by us, which reflect those that a market participant would use.
Our fair-value hierarchy for our financial assets measured at fair value on a recurring basis are as follows:
| December 31, 2021 | |||||||||||
| Level 1 |
| Level 2 |
| Level 3 |
| Total | |||||
(In thousands) | ||||||||||||
Assets: | ||||||||||||
Money-market funds classified as short-term investments | $ | | $ | — | $ | — | $ | | ||||
Money-market funds classified as non-current restricted investments |
| |
| — |
| — |
| | ||||
Total | $ | | $ | — | $ | — | $ | | ||||
| December 31, 2020 | |||||||||||
| Level 1 |
| Level 2 |
| Level 3 |
| Total | |||||
(In thousands) | ||||||||||||
Assets: |
|
|
|
|
|
|
| |||||
Money-market funds classified as short-term investments | $ | | $ | — | $ | — | $ | | ||||
Money-market funds classified as non-current restricted investments |
| |
| — |
| — |
| | ||||
Total | $ | | $ | — | $ | — | $ | | ||||
Cash held in demand deposit accounts of $
See “Note 9--Unsecured Convertible Senior Notes” for the carrying amount and estimated fair value of our
74
Note 7—Certain Balance Sheet Accounts
Property and Equipment, Net
Property and equipment, net consists of the following:
| December 31, |
| December 31, | |||
2021 | 2020 | |||||
(In thousands) | ||||||
Finance leases | $ | | $ | | ||
Laboratory equipment |
| |
| | ||
Computer equipment |
| |
| | ||
Office equipment and furniture |
| |
| | ||
Total cost |
| |
| | ||
Less accumulated depreciation and amortization |
| ( |
| ( | ||
Total property and equipment, net | $ | | $ | | ||
For the years ended December 31, 2021, 2020 and 2019, depreciation and amortization expenses were $
Accrued Expenses
Accrued expenses consist of the following:
| December 31, |
| December 31, | |||
2021 | 2020 | |||||
(In thousands) | ||||||
Sales rebates, fees and discounts | $ | | $ | | ||
Consulting and professional fees |
| | | |||
Interest payable |
| |
| | ||
Contract research and development | | | ||||
Employee compensation |
| |
| | ||
Clinical trials |
| |
| | ||
Other accrued expenses |
| |
| | ||
Total accrued expenses | $ | | $ | | ||
Note 8—Line of Credit
We have a Loan and Security Agreement with Silicon Valley Bank (“SVB”), which provides for a $
75
Interest on amounts outstanding is payable monthly at a floating rate equal to the greater of
The Line of Credit Agreement includes customary events of default that include, among other things, breach, non-payment, inaccuracy of representations and warranties, the occurrence of a material adverse change in our business or prospects for repayment of the Line of Credit Agreement, cross default to material indebtedness or material agreements, bankruptcy and insolvency, material judgments and a change in control. In the event of default, SVB may require all obligations under the Line of Credit Agreement to be immediately due and payable and charge a default rate of interest thereon. Additionally, under the loan and security agreement with SVB, we have agreed not to pay any dividends.
As of December 31, 2021 and 2020, we had
Note 9—Unsecured Convertible Senior Notes
On January 1, 2021, we adopted ASU 2020-06, Debt—Debt with Conversion Options (Subtopic 470-20) and Derivatives and Hedging—Contracts in Entity’s Own Equity (Subtopic 815-40) on a modified retrospective basis. ASU 2020- removes the separate liability and equity accounting for our convertible senior notes. Consequently, we now account for our convertible senior notes wholly as debt. Upon adoption, we removed the equity component allocated to debt issuance costs increasing unsecured convertible senior notes and shareholders’ equity by $
In November 2018, we issued $
76
Unsecured convertible senior notes outstanding at December 31, 2021 and 2020, respectively, are as follows:
Balance as of December 31, 2021 | |||||||||
| 2023 Notes |
| 2026 Notes |
| Total | ||||
(In thousands) | |||||||||
Principal amount | $ | | $ | | $ | | |||
Unamortized debt issuance costs |
| ( |
| ( |
| ( | |||
Total unsecured convertible senior notes, net | $ | | $ | | $ | | |||
Fair value of outstanding unsecured convertible senior notes (1) | $ | | $ | | |||||
Amount by which the unsecured convertible senior notes if-converted value exceeds their principal amount | $ | — | $ | — | |||||
Balance as of December 31, 2020 | |||||||||
| 2023 Notes |
| 2026 Notes |
| Total | ||||
(In thousands) | |||||||||
Principal amount | $ | | $ | | $ | | |||
Unamortized discount |
| ( |
| ( |
| ( | |||
Unamortized issuance costs attributable to liability component |
| ( |
| ( |
| ( | |||
Total unsecured convertible senior notes, net | $ | | $ | | $ | | |||
Fair value of outstanding unsecured convertible senior notes (1) | $ | | $ | | |||||
Amount by which the unsecured convertible senior notes if-converted value exceeds their principal amount | $ | | $ | | |||||
Equity component | $ | | $ | | |||||
Unamortized issuance costs |
| ( |
| ( | |||||
Net carrying amount of equity component (2) | $ | | $ | | |||||
| (1) | The fair value is classified as Level 3 due to the limited trading activity for the unsecured convertible senior notes. |
| (2) | Included in the consolidated balance sheet within additional paid-in capital. |
2023 Convertible Senior Notes
In November 2018, we issued $
The 2023 Notes are convertible into cash, shares of our common stock or a combination thereof, as we elect at our sole discretion. The initial conversion rate is
In August and September 2020, we issued the 2026 Notes and used approximately $
77
The settlement consideration was allocated between the repurchase of the liability and the equity component with the fair value of the liability component estimated to be $
In connection with the repurchase of $
The following table sets forth total interest expense recognized in connection with the 2023 Notes:
Year Ended December 31, | ||||||||
2021 |
| 2020 |
| 2019 | ||||
(In thousands) | ||||||||
Contractual interest expense | $ | | $ | | $ | | ||
Amortization of debt issuance costs |
| |
| |
| | ||
Amortization of debt discount |
| — |
| |
| | ||
Total | $ | | $ | | $ | | ||
78
2026 Convertible Senior Notes
In August and September 2020, we issued $
| (In thousands) | ||
2026 Notes principal amount issued | $ | | |
Repurchase of 2023 Notes |
| ( | |
Purchase of 2026 Capped Call |
| ( | |
Termination of the 2023 Capped Call contracts related to debt repurchased |
| | |
Issuance costs |
| ( | |
Net proceeds available for corporate use | $ | | |
The 2026 Notes are unsecured and accrue interest at an annual rate of
The initial conversion rate is
The 2026 Notes are convertible at the option of the holders on or after November 15, 2025 at any time prior to the close of business on February 12, 2026, the second scheduled trading day immediately before the stated maturity date of February 15, 2026. Additionally, holders may convert their 2026 Notes at their option at specified times prior to the maturity date only if:
| (1) | during any calendar quarter, beginning after September 30, 2020, that the last reported sale price per share of our common stock exceeds |
| (2) | during the consecutive business days immediately after any -consecutive-trading-day period (such -consecutive-trading-day period, the “measurement period”) in which the trading price per $ |
| (3) | there is an occurrence of one or more certain corporate events or distributions of our common stock; or |
| (4) | we call the 2026 Notes for redemption. |
We may elect, at our sole discretion, to convert the 2026 Notes into cash, shares of our common stock or a combination thereof.
Subject to the satisfaction of certain conditions, we may redeem in whole or in part the 2026 Notes at our option beginning August 15, 2023 through the 50th scheduled trading day immediately before the maturity date at a cash redemption price equal to the principal amount of the 2026 Notes to be redeemed plus any accrued and unpaid interest to, but excluding, the redemption date. The 2026 Notes are subject to redemption only if certain requirements are satisfied, including that the last reported sale price per share of our common stock exceeds
79
In order to reduce the dilutive impact or potential cash expenditure associated with the conversion of the 2026 Notes, we entered into capped call transactions in connection with the issuances of the 2026 Notes (the 2026 Capped Call). The 2026 Capped Call will cover, subject to anti-dilution adjustments substantially similar to those applicable to the 2026 Notes, the number of shares of common stock underlying the 2026 Notes when our common stock is trading within the range of approximately $
We evaluated the accounting for the issuance of the 2026 Notes and concluded that the embedded conversion features meet the requirements for a derivative scope exception for instruments that are both indexed to an entity’s own stock and classified in stockholders’ equity in its balance sheet, and that the cash conversion guidance applies. Therefore, proceeds of $
Further, we concluded the 2026 Capped Call qualifies for a derivative scope exception for instruments that are both indexed to an entity’s own stock and classified in stockholders’ equity in its balance sheet. Consequently, the fair value of the 2026 Capped Call of $
In accounting for the issuance of the 2026 Notes, we separated the 2026 Notes into liability and equity components, using an effective interest rate of
The following table sets forth interest expense recognized related to the 2026 Notes:
Year Ended December 31, | ||||||||
2021 |
| 2020 |
| 2019 | ||||
(In thousands) | ||||||||
Contractual interest expense | $ | | $ | | $ | — | ||
Amortization of debt issuance costs |
| |
| |
| — | ||
Amortization of debt discount |
| — |
| |
| — | ||
Total | $ | | $ | | $ | — | ||
Future minimum principal for the 2023 and 2026 Notes as of December 31, 2021 are as follows:
| (In thousands) | ||
2022 |
| $ | — |
2023 |
| | |
2024 |
| — | |
2025 |
| — | |
2026 |
| | |
Total future minimum principal payments under the convertible senior notes |
| $ | |
Note 10—Lease Liabilities
We have operating leases related to our office and laboratory space. The initial term of the leases is through November 2027 and we have
80
Lease-related assets and liabilities recorded on the balance sheet are as follows:
December 31, | December 31, | |||||
2021 |
| 2020 | ||||
(In thousands) | ||||||
Assets | ||||||
Operating lease assets | $ | |
| $ | | |
Finance lease assets, net |
| |
| | ||
Total lease assets | $ | |
| $ | | |
Liabilities | ||||||
Current: | ||||||
$ | |
| $ | | ||
| |
| | |||
Non-current: | ||||||
| |
| | |||
| |
| | |||
Total lease liabilities | $ | |
| $ | | |
Weighted-average remaining lease term | ||||||
Operating leases |
|
| ||||
Finance leases |
|
| ||||
Weighted-average discount rate | ||||||
Operating leases |
| | % |
| | % |
Finance leases |
| | % |
| | % |
The components of total lease costs are as follows:
Year Ended | |||||
December 31, | |||||
2021 |
| 2020 | |||
(In thousands) | |||||
Lease cost |
|
|
| ||
Operating lease cost | $ | | $ | | |
Finance lease cost: |
|
|
| ||
Amortization |
| |
| | |
Interest |
| |
| | |
Variable lease cost |
| |
| | |
Sublease income |
| ( |
| ( | |
Net lease cost | $ | | $ | | |
81
The supplemental cash flow information related to leases during 2021 is as follows:
Year Ended | |||||
December 31, | |||||
2021 |
| 2020 | |||
(In thousands) | |||||
Cash paid for amounts included in the measurement of lease liabilities | |||||
Cash payments for operating leases | $ | | $ | | |
Cash payments for financing leases | $ | |
| $ | |
The future maturities of our lease liabilities as of December 31, 2021 are as follows:
Operating | Finance | |||||
| Leases |
| Leases | |||
(In thousands) | ||||||
2022 | $ | | $ | | ||
2023 |
| |
| | ||
2024 |
| |
| | ||
2025 |
| |
| — | ||
2026 |
| |
| — | ||
Thereafter |
| |
| — | ||
Total undiscounted lease payments | | | ||||
Less interest | ( | ( | ||||
Total lease liabilities | $ | | $ | | ||
In January 14, 2022, we entered into an agreement with our landlord to early terminate a portion of the rentable square footage of our office and lab premises. Effective December 31, 2021, the square footage was reduced by
Note 11—Commitments and Contingencies
Contracts
We have various agreements with third parties that collectively require payment of termination fees totaling $
Development Milestones and Product Royalties
We have licensed a variety of intellectual property from third parties that we are currently developing or may develop in the future. These licenses may require milestone payments during the clinical development processes or upon approval of commercial sale as well as low single to low double-digit royalties on the net income or net sales of the product. For the years ended December 31, 2021 and December 31, 2020, we paid $
82
Note 12—Shareholders’ Equity
Common Stock
As of December 31, 2021, we had reserved shares of common stock under our equity plans as follows:
Options granted and outstanding |
| |
Restricted stock units granted and outstanding | | |
Common stock warrants | | |
Awards available under issuance under the 2017 Plan |
| |
Total shares reserved |
| |
Securities Offerings – In August 2020, we sold
In December 2019, we sold
At the Market Sales Agreement – We have a sales agreement to sell shares of our common stock having an aggregate offering price of up to $
Warrants
In connection with various previously outstanding debt agreements we have issued warrants to purchase shares of our common stock as follows:
Outstanding At |
|
| |||
December 31, 2021 | Expiration Date | Exercise Price | |||
April 12, 2023 | $ | | |||
Note 13—Stock-Based Compensation
Our equity plans provide for the grant of incentive and non-statutory stock options, stock appreciation rights, restricted stock awards, restricted stock units, performance units, performance shares and other stock and cash awards to employees, directors and consultants. Stock options are granted with an exercise price not less than the fair market value of Omeros’ common stock on the date of the grant. Any unexercised options expire
Vesting schedules for our equity plans generally are as follows:
Grant Type |
| Vesting Schedule |
Employee initial options grants |
| |
Employee recurring options grants |
| monthly |
Board member initial options grants |
| |
Board member recurring options grants |
| |
Non-employee consultant options grants |
| or monthly |
Employee RSUs |
In November 2020, restricted stock awards (“RSA’s”) totaling
83
In November 2021, RSA’s totaling
In December 2021, the Company granted
Stock-based compensation expense is as follows:
Year Ended December 31, | ||||||||
2021 |
| 2020 |
| 2019 | ||||
(In thousands) | ||||||||
Continuing operations | ||||||||
Research and development | $ | | $ | | $ | | ||
Selling, general and administrative |
| |
| |
| | ||
Total stock-based compensation in continuing operations | | | | |||||
Discontinued operations | | | | |||||
Total Stock-based compensation | $ | | $ | | $ | | ||
The fair value of each option grant is estimated on the date of grant using the Black-Scholes option-pricing model. The following assumptions were applied to stock option grants during the periods ended:
Year Ended December 31, |
| ||||||||
2021 | 2020 | 2019 | |||||||
Estimated weighted-average fair value | $ | | $ | | $ | | |||
Weighted-average assumptions: |
|
|
|
|
| ||||
Expected volatility |
| | % |
| | % |
| | % |
Expected life, in years |
|
|
| ||||||
Risk-free interest rate |
| | % |
| | % |
| | % |
Expected dividend yield |
| | % |
| | % |
| | % |
Expected volatility is based on the historical volatility of our stock price weighted by grant issuances over the reporting period. We use the simplified method to calculate expected life used in the valuation of our stock options. The risk-free interest rate is based on the U.S. Treasury yield curve in effect at the time of grant. Forfeiture expense is estimated at the time of grant and revised in subsequent periods if actual forfeitures differ from those estimates.
Stock option activity for all stock plans is as follows:
|
| Weighted- |
|
| ||||||
Average | Aggregate | |||||||||
Exercise | Remaining | Intrinsic | ||||||||
Options | Price per | Contractual Life | Value | |||||||
Outstanding | Share | (In years) | (In thousands) | |||||||
Balance at December 31, 2020 |
| | $ | |
|
|
|
| ||
Granted |
| |
| |
|
|
|
| ||
Exercised |
| ( |
| |
|
|
|
| ||
Forfeited |
| ( |
| |
|
|
|
| ||
Balance at December 31, 2021 |
| | $ | |
| $ | | |||
Vested and expected to vest at December 31, 2021 |
| | $ | |
| $ | | |||
Exercisable at December 31, 2021 |
| | $ | |
| $ | | |||
The total intrinsic value of options exercised during the years ended December 31, 2021, 2020 and 2019 was $
84
At December 31, 2021, there were
Note 14—Income Taxes
The components of income tax benefit from continuing operations are as follows:
| December 31, | ||||||||
| 2021 |
| 2020 |
| 2019 | ||||
(In thousands) | |||||||||
Current income tax expense: |
|
|
|
|
| ||||
Federal | $ | | $ | | $ | | |||
State |
| |
| |
| | |||
Total current income tax expense |
| |
| |
| | |||
Deferred income tax expense (benefit) |
|
|
|
|
|
| |||
Federal |
| |
| ( |
| ( | |||
State |
| |
| ( |
| ( | |||
Total deferred income tax expense (benefit) |
| |
| ( |
| ( | |||
Income tax expense (benefit) | $ | | $ | ( | $ | ( | |||
In December 2019, the Financial Accounting Standards Board issued ASU 2019-12, Income Taxes (Topic 740), which is intended to simplify various aspects of the income tax accounting guidance. ASU 2019-12 eliminates the exception to the incremental approach of intra-period tax allocation when there is a loss from continuing operations and income or gain from other items. As the Company prospectively adopted ASU 2019-12 January 1, 2021, we did not apply any intraperiod allocation rules to 2021.
To reflect intra-period tax allocation rules in prior years, we reclassified the tax benefit of income from discontinued operations to offset losses from continuing operations. During 2020, we recorded an income tax benefit of $
Under intraperiod allocation rules, the deferred tax liability related to the convertible debt and income earned from the sale of assets to Rayner, is a source of income that can be used to recognize the tax benefit of the current year loss through continuing operations. Deferred income taxes reflect the tax effect of net operating loss and tax credit carryforwards and the net temporary difference between the carrying amounts of assets and liabilities for financial reporting purposes and the amounts used for income tax purposes.
For the year ended December 31, 2021, we recorded state income tax expense of $
85
our consolidated balance sheet as of December 31, 2021 and $
Significant components of deferred income taxes are as follows:
| December 31, | |||||
| 2021 |
| 2020 | |||
(In thousands) | ||||||
Deferred tax assets: |
|
|
| |||
Net operating loss carryforwards | $ | | $ | | ||
Research and development tax credits |
| |
| | ||
Stock-based compensation |
| |
| | ||
Lease liability |
| |
| | ||
Disallowed interest expense | — | | ||||
Other |
| |
| | ||
Total deferred tax assets |
| |
| | ||
Deferred tax liabilities: |
|
|
|
| ||
Property and equipment | ( | ( | ||||
Gain on discontinued operations | ( | — | ||||
Equity component of Convertible Notes |
| — |
| ( | ||
Right of use assets | ( | ( | ||||
Total deferred tax liabilities |
| ( |
| ( | ||
Net deferred tax assets before valuation allowance |
| |
| | ||
Less valuation allowance |
| ( |
| ( | ||
Net deferred tax liabilities | $ | ( | $ | | ||
Net deferred tax liabilities are are included as other accrued liabilities – noncurrent in our consolidated balance sheet as of December 31, 2021.
As of December 31, 2021 and 2020, we had federal net operating loss carryforwards of approximately $
In certain circumstances, due to ownership changes, our net operating loss and tax credit carryforwards may be subject to limitations under Section 382 of the Internal Revenue Code. To date, we have not completed a Section 382 study. Unless previously utilized, net operating losses of $
We have established a
86
Reconciliation of income tax computed at federal statutory rates to the reported provisions for income taxes on continuing operations is as follows:
| Year ended December 31, |
| |||||
| 2021 |
| 2020 |
| 2019 |
| |
U.S. Federal statutory rate on net loss | ( | % | ( | % | ( | % | |
State tax, net of federal tax benefit | ( | % | ( | % | ( | % | |
Change in valuation allowance | | % | | % | | % | |
Tax credits | ( | % | ( | % | ( | % | |
Other | | % | ( | % | | % | |
Effective tax rate | ( | % | ( | % | ( | % | |
We file federal and certain state income tax returns, which provides varying statutes of limitations on assessments. However, because of net operating loss carryforwards, substantially all our tax years remain open to federal and state tax examination.
We recognize interest and penalties related to the underpayment of income taxes as a component of income tax expense. To date, there have been no interest or penalties charged to us in relation to the underpayment of income taxes.
On March 27, 2020, the Coronavirus Aid, Relief, and Economic Security Act (CARES Act) was enacted and signed into law in response to COVID-19. The CARES Act, among other things, includes several significant provisions which impact corporate taxpayers' accounting for income taxes, including a modification to the utilization of net operating losses and interest expense deduction limitations. The provisions of the CARES Act do not impact our tax provision.
Note 15—401(k) Retirement Plan
Our 401(k) retirement plan provides for an annual company discretionary match on employee contributions up to
87
ITEM 9. CHANGES IN AND DISAGREEMENTS WITH ACCOUNTANTS ON ACCOUNTING AND FINANCIAL DISCLOSURE
None.
ITEM 9A. CONTROLS AND PROCEDURES
Disclosure Controls and Procedures
Our management, with the participation of our principal executive officer and principal financial officer, evaluated the effectiveness of our disclosure controls and procedures, as defined in Rules 13a-15(e) and 15d-15(e) under the Exchange Act, as of December 31, 2021. Management recognizes that any controls and procedures, no matter how well designed and operated, can provide only reasonable assurance of achieving their objectives, and management necessarily applies its judgment in evaluating the cost-benefit relationship of possible controls and procedures. Based on the evaluation of our disclosure controls and procedures as of December 31, 2021, our principal executive and principal financial officers concluded that, as of such date, our disclosure controls and procedures were effective at the reasonable assurance level.
Management’s Report on Internal Control Over Financial Reporting
Our management is responsible for establishing and maintaining adequate internal control over financial reporting, as such term is defined in Exchange Act Rules 13a-15(f) and 15d-15(f). Internal control over financial reporting is a process designed to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles.
Internal control over financial reporting cannot provide absolute assurance of achieving financial reporting objectives because of its inherent limitations. Internal control over financial reporting is a process that involves human diligence and compliance and is subject to lapses in judgment and breakdowns resulting from human failures. Internal control over financial reporting also can be circumvented by collusion or improper management override. Because of such limitations, there is a risk that material misstatements may not be prevented or detected on a timely basis by internal control over financial reporting. However, these inherent limitations are known features of the financial reporting process. Therefore, it is possible to design into the process safeguards to reduce, though not eliminate, this risk.
Our management, with the participation of our principal executive and principal financial officers, conducted an assessment of the effectiveness of our internal control over financial reporting as of December 31, 2021. In making this assessment, management used the criteria set forth by the Committee of Sponsoring Organizations of the Treadway Commission in Internal Control-Integrated Framework (2013 framework). Based on the results of this assessment and on those criteria, our management concluded that our internal control over financial reporting was effective as of December 31, 2021.
Ernst & Young LLP has independently assessed the effectiveness of our internal control over financial reporting as of December 31, 2021 and its report is included below.
There was no change in our internal control over financial reporting identified in connection with the evaluation required by Rules 13a-15(d) and 15d-15(d) of the Exchange Act that occurred during our fourth fiscal quarter of 2021 that has materially affected, or is reasonably likely to materially affect, our internal control over financial reporting.
88
Report of Independent Registered Public Accounting Firm
To the Shareholders and the Board of Directors
Omeros Corporation
Opinion on Internal Control Over Financial Reporting
We have audited Omeros Corporation’s internal control over financial reporting as of December 31, 2021, based on criteria established in Internal Control—Integrated Framework issued by the Committee of Sponsoring Organizations of the Treadway Commission (2013 framework), (the COSO criteria). In our opinion, Omeros Corporation (the Company) maintained, in all material respects, effective internal control over financial reporting as of December 31, 2021, based on the COSO criteria.
We also have audited, in accordance with the standards of the Public Company Accounting Oversight Board (United States), the consolidated balance sheets of Omeros Corporation as of December 31, 2021 and 2020, the related consolidated statements of operations and comprehensive loss, shareholders' equity (deficit) and cash flows for each of the three years in the period ended December 31, 2021, and the related notes and our report dated March 1, 2022 expressed an unqualified opinion thereon.
Basis for Opinion
The Company’s management is responsible for maintaining effective internal control over financial reporting and for its assessment of the effectiveness of internal control over financial reporting included in the accompanying Management’s Annual Report on Internal Control Over Financial Reporting. Our responsibility is to express an opinion on the Company’s internal control over financial reporting based on our audit. We are a public accounting firm registered with the PCAOB and are required to be independent with respect to the Company in accordance with the U.S. federal securities laws and the applicable rules and regulations of the Securities and Exchange Commission and the PCAOB.
We conducted our audit in accordance with the standards of the PCAOB. Those standards require that we plan and perform the audit to obtain reasonable assurance about whether effective internal control over financial reporting was maintained in all material respects. Our audit included obtaining an understanding of internal control over financial reporting, assessing the risk that a material weakness exists, testing and evaluating the design and operating effectiveness of internal control based on the assessed risk, and performing such other procedures as we considered necessary in the circumstances. We believe that our audit provides a reasonable basis for our opinion.
Definition and Limitations of Internal Control Over Financial Reporting
A company’s internal control over financial reporting is a process designed to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles. A company’s internal control over financial reporting includes those policies and procedures that (1) pertain to the maintenance of records that, in reasonable detail, accurately and fairly reflect the transactions and dispositions of the assets of the company; (2) provide reasonable assurance that transactions are recorded as necessary to permit preparation of financial statements in accordance with generally accepted accounting principles, and that receipts and expenditures of the company are being made only in accordance with authorizations of management and directors of the company; and (3) provide reasonable assurance regarding prevention or timely detection of unauthorized acquisition, use, or disposition of the company’s assets that could have a material effect on the financial statements.
Because of its inherent limitations, internal control over financial reporting may not prevent or detect misstatements. Also, projections of any evaluation of effectiveness to future periods are subject to the risk that controls may become inadequate because of changes in conditions, or that the degree of compliance with the policies or procedures may deteriorate.
/s/ Ernst & Young LLP |
Seattle, Washington
March 1, 2022
89
ITEM 9B. OTHER INFORMATION
None
ITEM 9C. DISCLOSURE REGARDING FOREIGN JURISDICTIONS THAT PREVENT INSPECTIONS
Not Applicable.
PART III
ITEM 10. DIRECTORS, EXECUTIVE OFFICERS AND CORPORATE GOVERNANCE
The information required by this item will be contained in our definitive proxy statement issued in connection with the 2022 Annual Meeting of Shareholders and is incorporated herein by reference. Certain information required by this item concerning executive officers is set forth in Part I of this Annual Report on Form 10-K under the heading “Business- Information About Our Executive Officers and Significant Employees.”
ITEM 11. EXECUTIVE COMPENSATION
The information required by this item will be contained in our definitive proxy statement issued in connection with the 2022 Annual Meeting of Shareholders and is incorporated herein by reference.
ITEM 12. SECURITY OWNERSHIP OF CERTAIN BENEFICIAL OWNERS AND MANAGEMENT AND RELATED SHAREHOLDER MATTERS
Except for the information set forth below, the information required by this item will be contained in our definitive proxy statement issued in connection with the 2022 Annual Meeting of Shareholders and is incorporated herein by reference.
Securities Authorized for Issuance Under Equity Compensation Plans
The following table provides certain information regarding our equity compensation plans in effect as of December 31, 2021:
|
| Weighted- |
| Number of Securities | |||
Average | Remaining Available | ||||||
Number of Securities | Exercise Price | for | |||||
to be | of | Future Issuance | |||||
Issued Upon Exercise | Outstanding | Under | |||||
of | Options, | Equity | |||||
Outstanding Options, | Warrants and | Compensation | |||||
Warrants and Rights | Rights | Plans | |||||
Equity compensation plans approved by security holders: |
|
|
|
|
|
| |
2017 Omnibus Incentive Compensation Plan (1) |
| 6,766,499 | $ | 9.66 |
| 6,046,652 | |
2008 Equity Incentive Plan (2) |
| 5,943,388 | $ | 10.89 |
| — | |
Total |
| 12,709,887 | $ | 12.61 |
| 6,046,652 | |
| (1) | Our 2017 Omnibus Incentive Compensation Plan (the “2017 Plan”) provides for the grant of incentive and non-statutory stock options, restricted stock, restricted stock units, stock appreciation rights, performance units and performance shares to employees, directors and consultants and subsidiary corporations’ employees and consultants. The 2017 Plan replaced the Omeros Corporation 2008 Equity Incentive Plan (the “2008 Plan”), and as a result we will not grant any new awards under the 2008 Plan. Any stock option awards granted under the 2008 Plan that were outstanding as of the effective date of the 2017 Plan remain in effect pursuant to their terms and, if the award is canceled or is repurchased, the shares underlying such award become available for grant under the 2017 Plan. |
90
| (2) | The 2008 Plan provided for the grant of incentive and non-statutory stock options, restricted stock, stock appreciation rights, performance units and performance shares to employees, directors and consultants and subsidiary corporations’ employees and consultants. |
ITEM 13. CERTAIN RELATIONSHIPS AND RELATED TRANSACTIONS, AND DIRECTOR INDEPENDENCE
The information required by this item will be contained in our definitive proxy statement issued in connection with the 2022 Annual Meeting of Shareholders and is incorporated herein by reference.
ITEM 14. PRINCIPAL ACCOUNTING FEES AND SERVICES
The information required by this item will be contained in our definitive proxy statement issued in connection with the 2022 Annual Meeting of Shareholders and is incorporated herein by reference.
PART IV
ITEM 15. EXHIBITS, FINANCIAL STATEMENT SCHEDULES
1. Financial Statements
See the Index to Consolidated Financial Statements in Part II, Item 8 of this Form 10-K.
2. Financial Statement Schedules
All schedules have been omitted as the required information is either not required, not applicable or otherwise included in the Financial Statements and notes thereto.
3. Exhibits
The following list of exhibits includes exhibits submitted with this Form 10-K as filed with the SEC and those incorporated by reference to other filings
EXHIBIT INDEX
Incorporated by Reference | ||||||||||||
Exhibit |
| Exhibit Description |
| Form |
| File No. |
| Exhibit |
| Filing Date |
| Filed |
1.1 | Sales Agreement, dated March 1, 2021, between Omeros Corporation and Cantor Fitzgerald & Co. | 10-K | 001-34475 | 1.1 | 03/01/2021 | |||||||
3.1 | Amended and Restated Articles of Incorporation of Omeros Corporation | 10-K | 001-34475 | 3.1 | 03/31/2010 | |||||||
3.2 | 10-K | 001-34475 | 3.2 | 03/31/2010 | ||||||||
4.1 | 10-K | 001-34475 | 1.1 | 03/01/2021 | ||||||||
4.2 | S-1/A | 333-148572 | 4.1 | 10/02/2009 | ||||||||
91
4.3 | 8-K | 001-34475 | 10.3 | 05/19/2016 | ||||||||
4.4 | 8-K | 001-34475 | 10.2 | 4/13/2018 | ||||||||
4.5 | 8-K | 001-34475 | 4.1 | 11/15/2018 | ||||||||
4.6 | 8-K | 001-34475 | 4.1 | 08/14/2020 | ||||||||
4.7 | 8-K | 001-34475 | 4.2 | 08/14/2020 | ||||||||
10.1†† | X | |||||||||||
10.2* | S-1 | 333-148572 | 10.1 | 01/09/2008 | ||||||||
10.3* | 10-K | 001-34475 | 10.6 | 03/16/2017 | ||||||||
10.4* | Form of Stock Option Award Agreement under the 2008 Equity Incentive Plan | 10-Q | 001-34475 | 10.2 | 11/07/2013 | |||||||
10.5* | 2017 Omnibus Incentive Compensation Plan (as amended and restated effective as of June 11, 2021) | 8-K | 001-34475 | 10.1 | 6/16/2021 | |||||||
10.6* | Form of Stock Option Award Agreement under the 2017 Omnibus Incentive Compensation Plan | S-8 | 333-218882 | 4.4 | 06/21/2017 | |||||||
92
10.7* | 8-K | 001-34475 | 10.1 | 04/12/2010 | ||||||||
10.8* | S-1 | 333-148572 | 10.14 | 01/09/2008 | ||||||||
10.9* | S-1 | 333-148572 | 10.16 | 01/09/2008 | ||||||||
10.10* | Omeros Corporation Non-Employee Director Compensation Policy | 10-K | 001-34475 | 1.1 | 03/01/2021 | |||||||
10.11 | Lease dated January 27, 2012 between Omeros Corporation and BMR-201 Elliott Avenue LLC | 8-K | 001-34475 | 10.1 | 02/01/2012 | |||||||
10.12 | 10-Q | 001-34475 | 10.2 | 11/09/2012 | ||||||||
10.13 | 10-K | 001-34475 | 10.18 | 03/18/2013 | ||||||||
10.14 | 10-K | 001-34475 | 10.18 | 03/13/2014 | ||||||||
10.15 | 10-Q | 001-34475 | 10.3 | 11/09/2015 | ||||||||
10.16 | 10-Q | 001-34475 | 10.1 | 05/10/2017 | ||||||||
10.17 | 10-K | 001-34475 | 10.19 | 03/01/2019 | ||||||||
93
10.18 | 10-Q | 001-34475 | 10.1 | 08/08/2019 | ||||||||
10.19 | 10-K | 001-34475 | 10.20 | 03/02/2020 | ||||||||
10.20 | 10-Q | 001-34475 | 10.1 | 05/11/2020 | ||||||||
10.21 | 10-Q | 001-34475 | 10.1 | 11/09/2020 | ||||||||
10.22 | 10-K | 001-34475 | 1.1 | 03/01/2021 | ||||||||
10.23 | 10-K | 001-34475 | 1.1 | 03/01/2021 | ||||||||
10.24 | 10-Q | 001-34475 | 1.1 | 08/06/2021 | ||||||||
10.25† | S-1/A | 333-148572 | 10.29 | 09/16/2009 | ||||||||
10.26† | S-1 | 333-148572 | 10.30 | 01/09/2008 | ||||||||
10.27† | 10-K | 001-34475 | 10.24 | 03/16/2015 | ||||||||
94
10.28† | S-1/A | 333-148572 | 10.31 | 09/16/2009 | ||||||||
10.29† | S-1 | 333-148572 | 10.32 | 01/09/2008 | ||||||||
10.30† | S-1/A | 333-148572 | 10.47 | 09/16/2009 | ||||||||
10.31† | 10-K | 001-34475 | 10.28 | 03/18/2013 | ||||||||
10.32† | 10-Q | 001-34475 | 10.1 | 05/12/2010 | ||||||||
10.33† | 10-Q | 001-34475 | 10.1 | 05/10/2011 | ||||||||
10.34† | 10-Q | 001-34475 | 10.1 | 05/09/2013 | ||||||||
10.35† | Exclusive License Agreement between Omeros Corporation and Helion Biotech ApS dated April 20, 2010 | 10-Q | 001-34475 | 10.2 | 08/10/2010 | |||||||
10.36† | 10-K | 001-34475 | 10.44 | 03/15/2011 | ||||||||
10.37† | 10-K | 001-34475 | 10.45 | 03/15/2011 | ||||||||
95
10.38† | 10-K | 001-34475 | 10.46 | 03/16/2015 | ||||||||
10.39† | 10-Q | 001-34475 | 10.1 | 11/09/2015 | ||||||||
10.40 | 8-K | 001-34475 | 10.2 | 11/15/2018 | ||||||||
10.41 | 8-K | 001-34475 | 10.1 | 08/14/2020 | ||||||||
10.42 | 8-K | 001-34475 | 10.1 | 08/08/2019 | ||||||||
10.43 | 10-Q | 001-34475 | 10.1 | 08/10/2020 | ||||||||
10.44 | X | |||||||||||
10.45†† | 10-Q | 001-34475 | 10.1 | 11/12/2019 | ||||||||
23.1 | X | |||||||||||
31.1 | X | |||||||||||
96
31.2 | X | |||||||||||
32.1 | X | |||||||||||
32.2 | X | |||||||||||
101.INS | Inline XBRL Instance Document | X | ||||||||||
101.SCH | Inline XBRL Taxonomy Extension Schema Document | X | ||||||||||
101.CAL | Inline XBRL Taxonomy Extension Calculation Linkbase Document | X | ||||||||||
101.DEF | Inline XBRL Taxonomy Extension Definition Linkbase Document | X | ||||||||||
101.LAB | Inline XBRL Taxonomy Extension Labels Linkbase Document | X | ||||||||||
101.PRE | Inline XBRL Taxonomy Extension Presentation Linkbase Document | X | ||||||||||
104.1 | Cover Page Interactive Data File, formatted in Inline XBRL (included in Exhibit 101) | X |
* Indicates management contract or compensatory plan or arrangement.
† Portions of this exhibit are redacted in accordance with a grant of confidential treatment.
†† | Certain identified information has been excluded from the exhibit because it both (A) is not material and (B) would be competitively harmful if publicly disclosed. |
ITEM 16. FORM 10-K SUMMARY
Not included.
97
SIGNATURES
Pursuant to the requirements of the Securities Exchange Act of 1934, the registrant has duly caused this report to be signed on its behalf by the undersigned, thereunto duly authorized.
OMEROS CORPORATION | |
/s/ GREGORY A. DEMOPULOS, M.D. | |
Gregory A. Demopulos, M.D. | |
President, Chief Executive Officer |
Dated: March 1, 2022
Pursuant to the requirements of the Securities Exchange Act of 1934, this report has been signed below by the following persons on behalf of the registrant and in the capacities and on the dates indicated.
Signature |
| Title |
| Date |
/s/ GREGORY A. DEMOPULOS, M.D. | President, Chief Executive Officer and Chairman of the Board of Directors (Principal Executive Officer) | March 1, 2022 | ||
Gregory A. Demopulos, M.D. | ||||
/s/ MICHAEL A. JACOBSEN | Vice President, Finance, Chief Accounting Officer and Treasurer (Principal Financial Officer and Principal Accounting Officer) | March 1, 2022 | ||
Michael A. Jacobsen | ||||
/s/ THOMAS F. BUMOL, PH.D. | Director | March 1, 2022 | ||
Thomas F. Bumol, Ph.D. | ||||
/s/ THOMAS J. CABLE | Director | March 1, 2022 | ||
Thomas J. Cable | ||||
/s/ PETER A. DEMOPULOS, M.D. | Director | March 1, 2022 | ||
Peter A. Demopulos, M.D. | ||||
/s/ ARNOLD C. HANISH | Director | March 1, 2022 | ||
Arnold C. Hanish | ||||
/s/ LEROY E. HOOD, M.D., PH.D. | Director | March 1, 2022 | ||
Leroy E. Hood, M.D., Ph.D. | ||||
/s/ RAJIV SHAH, M.D. | Director | March 1, 2022 | ||
Rajiv Shah, M.D. | ||||
/s/ KURT ZUMWALT | Director | March 1, 2022 | ||
Kurt Zumwalt |
98
Exhibit 10.1
ASSET PURCHASE AGREEMENT
By and Among
OMEROS CORPORATION
as Seller
RAYNER SURGICAL INC.
as Purchaser
and
solely for the purposes of Article V and Section 6.24
RAYNER SURGICAL GROUP LIMITED
as Parent Guarantor
Dated as of December 1, 2021
[***] CERTAIN IDENTIFIED INFORMATION HAS BEEN EXCLUDED FROM THE EXHIBIT BECAUSE IT BOTH (A) IS NOT MATERIAL AND (B) WOULD BE COMPETITIVELY HARMFUL IF PUBLICLY DISCLOSED
Table of contents
| Page | |
| | |
ARTICLE I Definitions | 1 | |
| | |
ARTICLE II Purchase and Sale | 4 | |
| | |
Section 2.1 | Purchased Assets. | 4 |
Section 2.2 | Excluded Assets | 5 |
Section 2.3 | Assumed Liabilities | 6 |
Section 2.4 | Retained Liabilities | 7 |
Section 2.5 | Non-Assignment; Consents | 8 |
Section 2.6 | The Closing | 10 |
Section 2.7 | Consideration | 11 |
Section 2.8 | Closing Deliverables | 17 |
Section 2.9 | Payment Mechanics. | 19 |
Section 2.10 | Withholding Taxes | 19 |
Section 2.11 | Indirect Taxes | 20 |
ARTICLE III Representations and Warranties of the Seller | 20 | |
Section 3.1 | Organization and Standing. | 20 |
Section 3.2 | Authority; Execution and Delivery; Enforceability. | 20 |
Section 3.3 | Non-Contravention and Approvals. | 21 |
Section 3.4 | Title; Sufficiency of Purchased Assets; Personal Property. | 22 |
Section 3.5 | Real Property | 22 |
Section 3.6 | Intellectual Property. | 22 |
Section 3.7 | Contracts. | 25 |
Section 3.8 | Taxes | 25 |
Section 3.9 | Litigation. | 25 |
Section 3.10 | Employee Matters. | 26 |
Section 3.11 | Compliance with Applicable Laws. | 27 |
Section 3.12 | Regulatory Compliance | 28 |
Section 3.13 | Brokers and Finders | 30 |
Section 3.15 | Solvency | 30 |
ARTICLE IV Representations and Warranties of Purchaser | 30 | |
Section 4.1 | Organization | 30 |
Section 4.2 | Authority; Execution and Delivery; Enforceability. | 30 |
Section 4.3 | Non-Contravention and Approvals. | 31 |
Section 4.4 | No Litigation | 31 |
Section 4.5 | Exclusivity of Representations; Independent Investigation. | 32 |
Section 4.6 | Solvency | 33 |
Section 4.7 | Brokers and Finders | 33 |
Section 4.8 | Debarment | 33 |
Section 4.9 | Financial Ability. | 33 |
| | |
ARTICLE V Representations and Warranties of Parent Guarantor | 34 | |
| | |
Section 5.1 | Organization | 34 |
i
Section 5.2 Authority; Execution and Delivery; Enforceability | 35 | |
| | |
ARTICLE VI Covenants | 35 | |
| | |
Section 6.1 | Conduct of Business. | 35 |
Section 6.2 | Access to Information. | 37 |
Section 6.3 | Confidentiality. | 39 |
Section 6.4 | Efforts; Antitrust Notifications. | 39 |
Section 6.5 | Services from Affiliates | 41 |
Section 6.6 | Publicity | 42 |
Section 6.7 | Use of Seller Names and Marks. | 42 |
Section 6.8 | Further Action | 43 |
Section 6.9 | Bulk Sales Law | 43 |
Section 6.10 | Litigation; Privileged Matters. | 43 |
Section 6.11 | Employee Matters. | 44 |
Section 6.12 | WARN Act | 47 |
Section 6.13 | Accounts Payable | 48 |
Section 6.14 | Wrong Pockets; Payments. | 48 |
Section 6.15 | Noncompetition | 48 |
Section 6.16 | Rights of the Parties During the Term of Certain Ancillary Agreements | 49 |
Section 6.17 | Quality Control. | 49 |
Section 6.18 | Insurance | 51 |
Section 6.19 | Trade Notification | 51 |
Section 6.20 | Transfer of Purchased Regulatory Approvals and Compliance with Regulatory Requirements. | 51 |
Section 6.21 | Exclusivity | 53 |
Section 6.22 | Financing. | 53 |
Section 6.23 | Financing Cooperation. | 55 |
Section 6.24 | Guaranty | 55 |
Section 6.25 | License | 56 |
Section 6.26 | Technology Transfer Payments | 57 |
Section 6.27 | Patents; Licensing; Royalty Buy-Out | 57 |
Section 6.28 | SVB Consent; UCC amendments | 57 |
Section 6.29 | Replicative Agreements | 58 |
| | |
ARTICLE VII Conditions to Closing | 58 | |
| | |
Section 7.1 | Conditions to Each Party’s Obligations | 58 |
Section 7.2 | Conditions to Obligation of Purchaser | 58 |
Section 7.3 | Conditions to Obligation of the Seller | 59 |
Section 7.4 | Frustration of Closing Conditions | 59 |
| | |
ARTICLE VIII Termination | 59 | |
| | |
Section 8.1 | Termination | 60 |
Section 8.2 | Effect of Termination. | 61 |
| | |
ARTICLE IX Indemnification; Survival | 61 | |
| | |
Section 9.1 | Indemnification by the Seller | 61 |
Section 9.2 | Indemnification by Purchaser | 62 |
ii
Section 9.3 | Indemnification Procedures. | 62 |
Section 9.4 | Limitations on Indemnification | 64 |
Section 9.5 | Calculation of Indemnity Payments. | 65 |
Section 9.6 | Exclusivity. | 66 |
Section 9.7 | Tax Treatment of Indemnification | 67 |
Section 9.8 | Survival | 67 |
| | |
ARTICLE X Tax matters | 67 | |
| | |
Section 10.1 | Tax Covenants. | 67 |
Section 10.2 | Cooperation | 68 |
Section 10.3 | Payment of Taxes and Tax Proceedings. | 69 |
| | |
ARTICLE XI Miscellaneous | 70 | |
| | |
Section 11.1 | Assignment | 70 |
Section 11.2 | No Third-Party Beneficiaries | 70 |
Section 11.3 | Fees and Expenses | 70 |
Section 11.4 | Notices | 71 |
Section 11.5 | Interpretation. | 71 |
Section 11.6 | Counterparts | 73 |
Section 11.7 | Entire Agreement. | 73 |
Section 11.8 | Severability | 73 |
Section 11.9 | Governing Law | 74 |
Section 11.10 | Waiver of Jury Trial | 74 |
Section 11.11 | Venue | 74 |
Section 11.12 | Amendments and Waivers | 74 |
Section 11.13 | Remedies; Specific Performance. | 75 |
Section 11.14 | Further Assurances | 76 |
Section 11.15 | Debt Financing Parties | 76 |
iii
Exhibits
Exhibit A | Form of Domain Name Assignment Agreement |
Exhibit B | Form of Patent Assignment Agreement |
Exhibit C | Form of Trademark Assignment Agreement |
Exhibit D | Terms of Transitional Services Agreement |
Exhibit E | Form of Transitional Trademark License Agreement |
Exhibit F | Assignment and Assumption Agreement |
Exhibit G | Form of Royalty Report |
Attachments
Seller Schedule
Purchaser Schedule
iv
ASSET PURCHASE AGREEMENT
This Asset Purchase Agreement (this “Agreement”), is entered into as of December 1, 2021 (the “Execution Date”), by and among Omeros Corporation, a Washington corporation (the “Seller”), Rayner Surgical Inc., a Delaware corporation (“Purchaser” and together with the Seller, each individually a “Party” and together, the “Parties”) and, solely for the purposes of Article V and Section 6.24, Rayner Surgical Group Limited, a company limited by shares incorporated under the laws of England (“Parent Guarantor”).
WHEREAS, the Seller and the Selling Affiliates are engaged in the Exploitation of the Product;
WHEREAS, the Seller desires to sell, assign, transfer or otherwise convey or cause the Selling Affiliates to sell, assign, transfer or otherwise convey to Purchaser, and Purchaser desires to purchase and acquire from the Seller or the Selling Affiliates, all of the Seller’s and the Selling Affiliates’ right, title and interest in and to the Purchased Assets, and, in connection therewith, Purchaser shall assume the Assumed Liabilities (collectively, the “Acquisition”); and
NOW, THEREFORE, in consideration of the premises, representations and warranties and covenants set forth herein, and for other good and valuable consideration, the receipt and sufficiency of which are hereby acknowledged, the Parties agree as set forth herein.
ARTICLE I
Definitions
“Accounts Receivable” means all accounts receivable, notes receivable and other indebtedness due and owed by any Third Party to Seller or any of its Affiliates in respect of any period prior to the Closing Date.
“Acquisition Proposal” means any proposal or offer from any Person (other than the Purchaser or its Affiliates) relating to any direct or indirect sale, license, disposition or acquisition of the Product or any of the Purchased Assets, other than sales of inventory in the ordinary course of business, in a single transaction or series of related transactions.
“Action” means any action, arbitration, hearing, complaint, litigation, suit, proceeding or government charge (whether civil, criminal or administrative).
“Affiliate” means, with respect to any Person, any other Person that directly or indirectly, through one or more intermediaries, controls, is controlled by, or is under common control with such Person, and, for the purposes of this definition, the term “control” (including the terms “controlled by” and “under common control with”) means the possession, directly or indirectly of the power to direct or cause the direction of the management and policies of such Person, whether through ownership of voting securities, by Contract or otherwise; provided, that unless expressly specified otherwise hereunder, in no event shall any member of the CVC Network be an Affiliate of Purchaser or Parent Guarantor and in no event shall Purchaser or Parent Guarantor be a member of the CVC Network.
1
“Ancillary Agreements” means the agreements and instruments to be executed and delivered in connection with this Agreement, including the Domain Name Assignment Agreement, the Patent Assignment Agreement, the Trademark Assignment Agreement, the Transitional Services Agreement, the Transitional Trademark License Agreement and the Assignment and Assumption Agreement.
“Anti-Corruption Laws” means all applicable anti-bribery and anti-corruption Laws, including the Foreign Corrupt Practices Act of 1977 (15 U.S.C. §§ 78dd-1 et seq.), the Bribery Act 2010, the federal Anti-Kickback Statute (42 U.S.C. § 1320a-7b(b)), and Laws enacted by member states and signatories implementing the OECD Convention Combating Bribery of Foreign Officials.
“Assumed Tax Liabilities” means (a) all liabilities for Taxes arising out of, in respect of or relating to the Business or the Purchased Assets for all Post-Closing Tax Periods, (b) Purchaser’s liability for Transfer Taxes pursuant to Section 10.1(b), and (c) liabilities for all Taxes in respect of or relating to the Business or the Purchased Assets resulting from a Purchaser Tax Act or attributable to any breach by Purchaser or any of its Affiliates of any covenant or other agreement hereunder.
“Books and Records” means all (a) promotional labeling, advertising, marketing materials, market research, Sales and Promotional Materials, pricing lists, consulting deliverables, materials and plans related to media, training (including any related outlines and quizzes/answers, if any), trade shows (including displays); and other related literature, catalogs, publications, videos and materials, including consumer, customer and end-user information, and (b) other books and records (other than the Regulatory Documentation) reasonably necessary to and used by Seller or the Selling Affiliates to Exploit the Product, in each case ((a) and (b)), that are (i) exclusively related to the Product or the Business and (ii) in Seller’s or any of the Selling Affiliates’ control and in the form currently maintained by Seller (e.g., electronic), and in all cases excluding any Seller Names and Marks. For clarity, Books and Records does not include (1) any general ledger or (2) books, documents, records, files or other items prepared in connection with or relating to the negotiation and consummation of the transactions contemplated by this Agreement or the Ancillary Agreements including any strategic, financial or Tax analyses relating to the divestiture of the Purchased Assets, the Assumed Liabilities, the Product and the Business.
“Business” means the Exploitation of the Product as conducted on or prior to the Execution Date by the Seller and the Selling Affiliates.
“Business Benefit Plan” means each employment, retirement, pension, deferred compensation, medical, dental, disability, life, severance, change-in-control, retention, vacation, incentive commission and bonus, fringe benefit, equity-based compensation, and stock purchase plan or program either (a) sponsored, maintained or contributed to or required to be maintained or contributed to by the Seller or any of its Affiliates for the benefit of any Business Employee or (b) under which the Seller or any Selling Affiliate could reasonably be expected to have any liability with respect to any Business Employee, in each case, (i) whether or not an “employee benefit plan” within the meaning of Section 3(3) of ERISA and (ii) excluding plans or programs required by applicable Law.
2
“Business Day” means any day other than Saturday, Sunday or a day on which banking institutions in New York, New York or London, United Kingdom are permitted or obligated by Law to remain closed.
“Business Employee” means any employee of the Seller or any of its Affiliates listed on Section 1.1(a) of the Seller Schedule as of the Execution Date (as such list may be updated from time to time prior to Closing upon mutual written agreement of Seller and Purchaser, the “Business Employee List”).
“Business Material Adverse Effect” means any event, change, occurrence, circumstance, condition, state of facts, development or effect that has had, or would reasonably be expected to have, individually or in the aggregate, (a) a material adverse effect on the business, results of operations or financial condition of the Purchased Assets, the Assumed Liabilities or the Business, taken as a whole, or (b) prevents, materially impedes, materially delays or is reasonably likely to prevent, materially impede or materially delay the consummation and performance by the Seller or its Affiliates of the transactions contemplated by this Agreement and the Ancillary Agreements, taken as a whole, and their respective obligations thereunder; provided, however, that none of the following, and no effect, change, event or occurrence arising out of or resulting from the following, shall constitute or be taken into account, individually or in the aggregate, in determining whether there has been or will be a Business Material Adverse Effect: (i) the Seller’s or any of its Affiliates’ compliance with the terms and conditions of this Agreement; (ii) any other action by the Seller or any of its Affiliates (1) expressly contemplated by this Agreement, (2) which Purchaser has expressly requested in writing or (3) to which Purchaser has consented in writing; (iii) any event, change, occurrence or effect affecting the industry, industry sectors or any geographic markets in which the Business operates generally or the United States or worldwide economy generally or the securities, syndicated loan, credit or other financial markets generally, including changes in interest or exchange rates; (iv) political or regulatory conditions, including the worsening of any existing conditions; (v) any natural disaster, health condition (including an epidemic, pandemic or disease outbreak, such as COVID-19), or any acts of terrorism, sabotage, military action or war (whether or not declared), or any escalation or worsening thereof, or any other force majeure event, whether or not caused by any Person, or any national or international calamity or crisis; (vi) any quarantine, “shelter in place,” “stay at home,” workforce reduction, social distancing, shut down, closure, sequester, safety or similar Law, directive, restrictions, guidelines, responses or recommendations of or promulgated by any Governmental Authority, including the Centers for Disease Control and Prevention and the World Health Organization, in each case, in connection with or in response to any health condition (including any epidemic, pandemic or disease outbreak, such as COVID-19) and any evolutions or mutations thereof or related or associated epidemics, pandemics or disease outbreaks (all of the foregoing, “Pandemic Measures”), including any change, effect, event or circumstance with respect to any health condition or Pandemic Measures or any escalation or worsening thereof (including any subsequent waves), (vii) any failure of the Seller or any of its Affiliates or the Business to meet internal or public forecasts, projections, predictions, guidance, estimates, milestones or budgets (provided, that the underlying reason for the failure to meet such forecasts, projections, predictions, guidance, estimates, milestones or budgets may be considered, except as otherwise excluded by this definition); (viii) the negotiation or execution of this Agreement or any Ancillary Agreement or the announcement or pendency or consummation of the Acquisition or a potential transaction involving the Business, including any litigation or
3
any loss of, or impact on the relationship of the Business with, any employees, partners, suppliers, customers, regulators or licensees; (ix) any acts or omissions of Purchaser or any of its Affiliates; (x) any use or market entry of any product that is a competitor to the Product (other than under a marketing authorization or other regulatory approval granted by a Governmental Authority to a Third Party); or (xi) any change or prospective change in Laws, US GAAP or other applicable accounting standards or the enforcement thereof; provided, further, that with respect to a matter described in any of clauses (iii), (iv), (v), (vi), and (xi), such event, change, occurrence or effect may be taken into account in determining whether there has been a Business Material Adverse Effect to the extent such event, change, occurrence or effect has a materially disproportionate adverse effect on the Business relative to other operating businesses similar to the Business.
“Calculation Time” means 11:59 p.m. (Eastern time) on the day immediately before the Closing Date.
“Code” means the Internal Revenue Code of 1986.
“Confidential Information” means (a) all information (whether prepared by the Seller, any of its Affiliates or otherwise) relating to the Seller, its Affiliates or their respective businesses, including the Business, including any technical, scientific, trade secret or other proprietary information with respect to the Seller or its Affiliates with which Purchaser or its representatives have or may come into contact in the course of their investigation, negotiation and consummation of the Acquisition and the other transactions contemplated hereby, whether before or after the Execution Date, and regardless of whether such information is furnished in writing, orally, in electronic form or otherwise and (b) all information related to the Seller or its Affiliates and their respective businesses, including the Business, contained in analyses, compilations, studies or other documents (regardless of the form in which any such analyses, compilations, studies or other documents are maintained) prepared by Purchaser or any of its representatives to the extent these contain or otherwise reflect any information referred to in clause (a) of this definition. Confidential Information shall not include any information which: (i) is or becomes publicly known or publicly available without any disclosure by Purchaser or any of its representatives or (ii) becomes lawfully available to Purchaser or any of its representatives on a non-confidential basis from a source other than the Seller or any of its Affiliates, which source is not under any obligation of confidentiality with respect to such information.
“Contract” means unexpired contracts, leases, subleases, licenses, agreements, guarantees, commitments, purchase orders, indentures, notes, bonds, loans or credit agreements, instruments, mortgages, deeds of trust license, powers of attorney, guaranties and all other legally binding written instruments, arrangements, understandings or obligations, in each case, as amended and supplemented from time to time and including all schedules, annexes and exhibits thereto and all renewals, replacements and substitutions therefor.
“Control” means, with respect to any Intellectual Property or Know-how, possession of the right, whether directly or indirectly and whether by ownership, license or otherwise, to grant a license, sublicense or other right to or under such Intellectual Property or Know-how as provided for herein without violating the terms of any Contract with any third party.
4
“Copyrights” means copyrights and applications and registrations and renewals therefor.
“COVID-19” means the disease caused by severe acute respiratory syndrome coronavirus novel coronavirus disease, COVID-19 virus (SARS-COV-2) (or related strains, variations, and sequences) or mutations, (or antigenic shifts) thereof or a public health emergency resulting therefrom, or associated epidemics, pandemics or disease outbreaks.
“CVC Funds” means funds or vehicles advised by *[***] and/or its Subsidiaries from time to time.
“CVC Network” means [***] and each of its Subsidiaries from time to time, (iii) the CVC Funds, (iv) any portfolio company invested in by the CVC Funds from time to time excluding Purchaser, Parent Guarantor and any other Persons through which the CVC Funds indirectly invest in Purchaser, and (v) [***] and each of its subsidiaries from time to time and any funds or entities managed or advised by them from time to time.
“Data Room” means the virtual data room maintained by the Seller or one or more of its representatives with Workspaces under the project name “Project Omaha” with respect to the transactions contemplated hereby.
“Diligent Efforts” means, with respect to the Subject Products, the performance of obligations or tasks in a manner consistent with diligent practices of companies of reasonably comparable size and resources in the pharmaceutical industry for the marketing, promotion and sale of a product having similar technical and regulatory characteristics and similar market potential, profit potential and strategic value, and that is at a similar stage in its product life cycle, in each case based on all relevant factors and all conditions then prevailing, and, solely with respect to the exercise of Diligent Efforts with respect to the Subject Products in the United States, [***]. For clarity, Diligent Efforts shall be assessed on a country-by-country basis.
“Domain Name Assignment Agreement” means that certain Domain Name Assignment Agreement to be entered into as of the Closing Date among Purchaser and the Seller or the Selling Affiliate(s) party thereto, for the assignment to Purchaser of the Seller’s and the Selling Affiliates’ right, title and interest to and in the Purchased Domain Names, substantially in the form of Exhibit A attached hereto.
“Domain Names” means registered internet domain names.
“Environmental Laws” means all applicable Laws and permits relating to pollution or the protection of the environment or natural resources, or human health and safety (as it relates to exposure to Hazardous Materials), or that classify, regulate, call for the remediation of, require reporting with respect to, or list or define air, water, groundwater, solid waste, hazardous or toxic substances, materials, wastes, pollutants or contaminants, or which regulate the manufacture,
* [***] CERTAIN IDENTIFIED INFORMATION HAS BEEN EXCLUDED FROM THE EXHIBIT BECAUSE IT BOTH (A) IS NOT MATERIAL AND (B) WOULD BE COMPETITIVELY HARMFUL IF PUBLICLY DISCLOSED
5
handling, transport, sale, distribution, reclamation, recycling, use, treatment, storage or disposal of, or exposure to, Hazardous Materials or materials containing Hazardous Materials.
“ERISA” means the Employee Retirement Income Security Act of 1974.
“ERISA affiliate” means, with respect to any entity, (a) a member of any “controlled group” (within the meaning of Section 414(b) of the Code) of which that entity is also a member, (b) a trade or business, whether or not incorporated, under common control (within the meaning of Section 414(c) of the Code) with that entity, or (c) a member of any affiliated service group (within the meaning of Section 414(m) of the Code) of which that entity is also a member.
“European Major Markets” means France, Germany, Italy, Spain and the United Kingdom.
“Excluded Contracts” means the Contracts related to the Business to which Seller or any of its Affiliates is a party that are not Purchased Contracts, as set forth on Section 1.1(b) of the Seller Schedule, in their entirety.
“Exploit” (and related terms such as “Exploitation”) means with respect to any product, the research, development (including seeking, obtaining or maintaining Regulatory Approvals), Manufacture, testing, packaging, labeling, storage, import, export, distribution, sale, licensing, commercialization, transportation, registration, outsourcing, advertising, marketing and promotion of such product, as applicable.
“FDA” means the United States Food and Drug Administration and any successor agency thereto.
“FDCA” means the Federal Food, Drug, and Cosmetic Act (21 U.S.C. § 301 et seq.) and all rules and regulations promulgated thereunder.
“Fraud” means an intentional misrepresentation of a representation or warranty in this Agreement or an Ancillary Agreement that constitutes common law fraud under the Laws of the State of Delaware.
“Generic Version” means, with respect to any Subject Product in a particular country, any pharmaceutical product that (a) is sold in such country by a Third Party that is not an Affiliate or licensee (regardless of tier) of Purchaser or any of its Affiliates under a marketing authorization or other regulatory approval granted by a Governmental Authority to a Third Party, (b) is a phenylephrine and ketorolac intraocular solution 1%/0.3% and (c) is approved in reliance on a prior Regulatory Approval of any Subject Product granted to Purchaser, any of Purchaser’s Affiliates or its or their licensees (regardless of tier) or assignees, or Seller or any of Seller’s Affiliates or its or their licensees (regardless of tier) or assignees by the applicable Governmental Authority; provided, however, that, for clarity, any Subject Product manufactured, sold or authorized by Purchaser, its Affiliates or (sub)licensees shall not constitute a Generic Version.
“Good Clinical Practice” or “GCP” means the standards for the clinical development and research of drugs, including all Laws and requirements relating to the protection of human subjects and the design, conduct, performance, monitoring, auditing, recording, analysis and
6
reporting of clinical trials, promulgated, enforced, or endorsed by any Governmental Authority, including 21 C.F.R. Parts 11, 50, 54, 56 and 312, the European Union’s Commission Directive 2005/28/EC, the corresponding national law of the European Union’s Member States and the United Kingdom’s Medicines for Human Use (Clinical Trials) Regulations 2004.
“Good Laboratory Practice” or “GLP” means the standards, practices, and procedures for good laboratory practices by research laboratories promulgated, enforced, or endorsed by any Governmental Authority, including 21 C.F.R. Part 58, the European Union’s Directive 2004/10/EC, the corresponding national law of the European Union’s Member States and the United Kingdom’s Good Laboratory Practice Regulations 1999.
“Good Manufacturing Practice” or “GMP” means the then-current applicable standards, practices and procedures for the methods to be used in, and the facilities or controls to be used for, the Manufacture of the Product or any Subject Product, as promulgated, enforced, or endorsed by any Governmental Authority, including FDA regulations at 21 C.F.R. Parts 210 and 211, and otherwise under 21 U.S.C. 351, the European Union’s Commission Directive 2003/94/EC, the corresponding national law of the European Union’s Member States and the United Kingdom’s Human Medicines Regulations 2012, or as otherwise required by applicable Laws, as in effect at the time of Manufacture.
“Governmental Authority” means any supranational, international, European Union, national, federal, state or local court, administrative agency or commission or other governmental authority or instrumentality, whether domestic or foreign, including FDA and any corresponding foreign agency, or any court or arbitral body, exercising executive, legislative, judicial, regulatory or administrative functions.
“Hazardous Material” means any substance, material or waste that is regulated, classified, or otherwise characterized under or pursuant to any Environmental Law as “hazardous,” “toxic,” “pollutant,” “contaminant,” “radioactive,” or words of similar meaning or effect, including petroleum and its by-products, asbestos, polychlorinated biphenyls, radon, mold and urea formaldehyde insulation.
“HSR Act” means the Hart-Scott-Rodino Antitrust Improvements Act of 1976.
“Indirect Taxes” means any and all sales, use, value added, goods and services, and similar turnover or gross margin Taxes incurred or imposed in respect of the transfer of the Business or the Purchased Assets pursuant to this Agreement; provided, however, that Washington B&O Tax is not an Indirect Tax.
“Intellectual Property” means the following: (a) Patents, (b) Trademarks and (c) Copyrights.
“Inventory Value Target Amount” means $700,000.
“Know-how” means any and all technical, scientific, commercial and other information, including inventions (whether patentable or not), trade secrets, know-how, confidential data, specifications, formulations, manufacturing processes, chemical or biological manufacturing control data, research and development data and quality control procedures and clinical data.
7
“Knowledge of the Purchaser” means the actual knowledge, after due inquiry, of any of the individuals listed on Section 1.1(c) of the Purchaser Schedule.
“Knowledge of the Seller” means the actual knowledge, after due inquiry, of any of the individuals listed on Section 1.1(c) of the Seller Schedule.
“Law” means any domestic or foreign, federal, state or local statute, law, treaty, judgment, ordinance, rule, administrative interpretation, regulation, order or other requirement having the force of law of any Governmental Authority.
“Liability” means, with respect to any Person, any direct or indirect liability, indebtedness, obligation, commitment, obligation, expense, claim, loss, deficiency, debt, guarantee or endorsement of or by such Person of any type, whether fixed, contingent or absolute, asserted or unasserted, matured or unmatured, liquidated or unliquidated, accrued or not accrued, known or unknown, due or to become due, whenever or however arising (including whether arising out of any Contract or tort based on negligence or strict liability).
“Liens” means any and all mortgages, liens, pledges or other encumbrances of any kind, including hypothecation, ownership interest of others, claim, easement, title retention agreement, voting trust agreement, option, right of first refusal, survey defect, imperfection of title, charge or other security interest.
“Manufacture” means all activities related to the design, production, manufacture, processing, filling, finishing, packaging, labeling, and shipping and holding (prior to distribution) of a pharmaceutical product or any intermediate quality assurance and quality testing thereof.
“Material Contracts” means (a) the Purchased Contracts and (b) all of the Contracts to which Seller or any Selling Affiliate is a party or by which it is bound that primarily relate to the Product or the Business or are necessary for the continued operation of the Business, including:
(i)any Contract (excluding purchase orders) for the manufacture or purchase of Products, active pharmaceutical ingredients or other materials, supplies, goods, equipment or other tangible assets in connection with the Business;
(ii)any sales, distribution, discounting or other similar agreement relating to the sale by Seller or its Affiliates of Products or other materials, supplies, goods, equipment or other tangible assets;
(iii)any partnership, joint venture, strategic alliance, collaboration, co-promotion, co-development, or research and development Contract with respect to the Product or the Business;
(iv)any agreement containing covenants expressly limiting or restricting in any material respect the freedom of the Seller or its Affiliates to compete in any line of business, therapeutic area or with any Person or in any geographic region and which would so limit or restrict the freedom of Buyer or its Affiliates with respect to the Business after the Closing Date;
8
(v)any agreement as obligor or guarantor relating to indebtedness or any other agreement mortgaging, pledging or otherwise placing a Lien (other than a Permitted Lien), any bank guarantees, performance or similar guarantees, in each case, with respect to the Purchased Assets;
(vi)any agreement that relates to research, clinical trial, development, distribution, license, marketing, promotion, commercialization, use, exploitation or manufacturing by any third party of the Product;
(vii)any agreement granting any right of first offer, right of first refusal or right of first negotiation with respect to any of the Purchased Assets;
(viii)any agreement that requires the applicable party thereto to purchase its total requirements of any product or services from the other party thereto in connection with the Business;
(ix)any agreement with a customer granting any “most favored nations” pricing terms that apply to the Business;
(x)any agreement that provides for the exclusive (A) manufacture of any element, (B) sale, or (C) distribution of the Product;
(xi)any agreement that imposes material minimum purchase obligations, including any obligation to purchase all or any portion of a party’s requirements for a product;
(xii)any agreement that contains material “take or pay” obligations;
(xiii)any agreement related to the settlement of any (i) material Action involving a claim for money damages, or (ii) any other material Action not involving a claim for money damages;
(xiv)any agreement to which any Governmental Authority is a party;
(xv)any agreements to which Seller or any of its Affiliates grants to or obtains from a third party a license (including sublicense) under any Intellectual Property, other than any (a) licenses for commercial off the shelf computer software that are generally available on nondiscriminatory pricing terms and (b) non-exclusive licenses granted to contractors in the ordinary course of business;
(xvi)Personal Property Leases;
(xvii)Contracts that are not terminable by Seller on notice of ninety (90) days or less without penalty;
(xviii)any acquisition or divestiture contract that contains financial covenants, indemnities or other payment obligations (including “earn-out,” “milestone” or other contingent payment obligations); or
9
(xix)any other agreement that, if terminated or not renewed (or if otherwise not in effect) would have a Business Material Adverse Effect.
“Milestone Event” means the occurrence of the event described on Section 1.1(d) of the Seller Schedule.
“Net Revenue” means, with respect to a Subject Product for any period, the gross amount billed or invoiced by or on behalf of the Purchaser, its Affiliates or its or their assignees or licensees (or sublicensees, regardless of tier) other than licensees (or sublicensees, regardless of tier) of rights to sell the applicable Subject Product in any country other than the United States, to Third Parties (including wholesalers or distributors) for the sale of such Subject Product, less deductions (without duplication) for: (a) normal and customary trade, quantity and prompt settlement discounts actually allowed; (b) amounts repaid or credited by reason of rejection, defects, return or recall of goods, rebates or bona fide price reductions (including wholesaler charge backs); (c) customs and excise duties and other Taxes or duties (excluding income Taxes) related to such sales (such as sales, value added, or use taxes) to the extent that such items are added to the sale price and set forth separately in the gross amount invoiced; and (d) price reductions or rebates and similar payments, retroactive or otherwise, imposed by, negotiated with or otherwise paid to any Governmental Authority such as, by way of illustration, federal or state Medicaid, Medicare or similar state program or equivalent governmental program of any jurisdiction outside of the United States; (e) the portion of administrative fees paid during the relevant time period to group purchasing organizations, pharmaceutical benefit managers or Medicare Prescription Drug Plans relating to such Subject Product; (f) freight, insurance, import/export, and other transportation charges to the extent added to the sale price and set forth separately as such in the total amount invoiced; and (g) any fees for services provided by wholesalers and warehousing chains related to the distribution of such Subject Product; provided, that the amounts deducted for such services under this clause (g) shall not exceed 5% of the gross amount billed or invoiced for such Subject Product in the relevant calendar quarter; provided, that any deductions listed in (a) through (g) that involves a payment by Purchaser, its Affiliates or its or their licensees (or sublicensees, regardless of tier) shall be taken as a deduction in the calendar quarter in which the payment is accrued by such entity in accordance with US GAAP. Any adjustments to the accruals set forth in (a) through (g) above that are made in future periods shall be included as an adjustment to Net Revenue in such future period.
The transfer of any unit of a Subject Product by Purchaser, its Affiliates or its or their licensees (or sublicensees, regardless of tier) to any of its Affiliates, licensees (or sublicensees, regardless of tier) shall not result in any Net Revenue unless such unit is consumed by such Affiliate, licensee (or sublicensees, regardless of tier) in the course of its commercial activities. In the case of pharmacy incentive programs, hospital performance incentive programs, chargeback claims, disease management programs, similar programs or discounts on portfolio product offerings, all rebates, discounts and other forms of reimbursements shall be allocated among products and/or services on the basis on which such rebates, discounts and other forms of reimbursements were actually granted or, if such basis cannot be determined, in accordance with the existing allocation methodology of Purchaser, its Affiliates or its or their licensees (or sublicensees, regardless of tier); provided, that any such allocation shall be done in accordance with applicable Law, including any price reporting Laws. Subject to the above, Net Revenue shall be calculated in accordance with US GAAP, consistently applied.
10
For purposes of this definition, “Combination Product” means any product that is comprised of or contains a Subject Product sold, for a single price, together with (a) one or more other active ingredients, either as a fixed dose product, co-formulated product or co-packaged product, or (b) one or more lens(es) or other medical device(s). Any delivery devices or systems, vehicles, adjuvants and excipients used in conjunction with any Subject Product shall not be treated as active ingredients or other medical devices for the purposes of this definition.
If any Subject Product is, or is sold as part of, a Combination Product in a given country, Net Revenue with respect to such Subject Product, shall be adjusted by multiplying by the fraction A/(A+B) where A is the gross amount per unit invoice price of the Subject Product in such country, if sold separately, and B is the gross amount per unit invoice price of any (i) other active ingredient(s) in the Combination Product in such country or (ii) lenses or other medical device(s) in the Combination Product in such country, in each case ((i) or (ii)), if sold separately. If, on a country-by-country basis, the other active ingredient(s) or lens(es) or other medical device(s), as applicable, in the Combination Product are not sold separately in that country, Net Revenue will be adjusted by multiplying by the fraction A/C where A is the gross amount per unit invoice price of the Subject Product in such country, if sold separately, and C is the gross amount per unit invoice price of the Combination Product in such country. In each case, the gross per unit invoice price shall be that applicable during the relevant calendar quarter or, if sales of both the Subject Product and the other product(s) or lens(es) or other medical device(s), as applicable did not occur in such calendar quarter, then in the most recent calendar quarter in which sales of both occurred. If neither the Subject Product nor the other active ingredient(s) or lens(es) or other medical device(s), as applicable, is sold in such country, the Parties shall discuss in good faith the adjustment for Net Revenue of such Combination Product, taking into consideration, in the such country, the relative value to the end user of each therapeutically active ingredient or lens or other medical device, as applicable, and the proportionality of pricing in other countries where the Subject Product and other active ingredient(s) or lens(es) or other medical device(s) are being sold separately, but in all cases excluding any Generic Version included therein.
“Patent(s)” means (a) all national, regional and international patents and patent applications, including provisional patent applications; (b) all patent applications filed either from such patents, patent applications or provisional applications or from an application claiming priority from either of these, including divisionals, continuations, continuations-in-part, provisionals, converted provisionals and continued prosecution applications; (c) all patents that have issued or in the future issue from the foregoing patent applications ((a) and (b)), including utility models, petty patents, innovation patents and design patents and certificates of invention; (d) all extensions or restorations by existing or future extension or restoration mechanisms, including revalidations, reissues, re-examinations and extensions (including any supplementary protection certificates and the like) of the foregoing patents or patent applications ((a), (b) and (c)); and (e) any similar rights, including so-called pipeline protection or any importation, revalidation, confirmation or introduction patent or registration patent or patent of additions to any of such foregoing patent applications and patents.
“Patent Assignment Agreement” means that certain Patent Assignment Agreement to be entered into as of the Closing Date among Purchaser and the Seller or the Selling Affiliate(s) party thereto, for the assignment to Purchaser of the Seller’s and the Selling Affiliates’ right, title
11
and interest to and in the Registered Intellectual Property that are Patents, substantially in the form of Exhibit B attached hereto.
“Permits” means any permits, licenses, franchises, registrations, variances, authorizations, accreditations, exemptions, consents, rights, privileges, certificates, orders and other approvals issued or granted by, or obtained from, any Governmental Authority, but for clarity Permits does not include Regulatory Approvals.
“Permitted Liens” means (a) such Liens as are set forth in Section 1.1(e) of the Seller Schedule, (b) mechanics’, carriers’, workmen’s, repairmen’s or other similar Liens which have arisen in the ordinary course of business, (c) Liens arising under original purchase price conditional sales Contracts and equipment leases with third parties entered into in the ordinary course of business, (d) Liens for Taxes and other governmental charges that are not yet delinquent or are being contested in good faith and for which adequate reserves have been recorded in accordance with US GAAP and (e) Liens which will be released on or prior to the Closing.
“Person” means any individual, firm, corporation, partnership, limited partnership, limited liability company, trust, joint venture, association, unincorporated organization, Governmental Authority or other entity.
“Post-Closing Tax Period” means any taxable period beginning after the Closing Date and the portion of any Straddle Tax Period beginning on the day after the Closing Date.
“Pre-Closing Tax Period” means any taxable period ending on or prior to the Closing Date and the portion of any Straddle Tax Period ending on the Closing Date.
“Product” means the pharmaceutical product known as Omidria®.
“Product Specifications” means the quality specifications for the Manufacture, testing, and release of the Product or the Subject Product, as specified, and their respective components.
“Purchased Domain Names” means the Domain Names set forth on Section 1.1(f) of the Seller Schedule.
“Purchased Equipment” means the tangible assets and properties (a) listed on Section 1.1(g) of the Seller Schedule or (b) acquired by the Seller or any Selling Affiliate between the Execution Date and the Closing Date in compliance with Section 6.1 that are used or held for use exclusively for the Business. For clarity, Purchased Equipment does not include any software or software licenses.
“Purchased Intellectual Property” means, collectively: (a) the Trademarks set forth on Section 1.1(h)(A) of the Seller Schedule, including all Trademark registrations and applications set forth thereon, all social media handles and all unregistered and common law rights in those Trademarks (“Purchased Trademarks”); (b) the issued Patents and Patent applications set forth on Section 1.1(h)(B) of the Seller Schedule; (c) the Purchased Know-how; (d) all other Intellectual Property exclusively related to the Product or the Business; and (e) any Actions commencing after the Closing Date relating to infringement thereof.
12
“Purchased Inventory” means all raw materials, API, work-in-process (i.e., naked vials) including PPQ lots that are expected to be useable for commercial purposes, finished goods, goods in transit (not including goods in transit to third party wholesalers which have been invoiced), and other materials and supplies used or held for use exclusively for the Business, including samples and packaging owned by Seller, in each case, to the extent useable or saleable, as applicable, in the ordinary course.
“Purchased Know-how” means all Know-how that is owned by the Seller or any of the Selling Affiliates and that is exclusively related to the Product or the Business, including any Action commencing after the Closing Date for misappropriation thereof.
“Purchased Prepaid Items” means all credits, prepaid expenses, deferred charges as accounted for under US GAAP, advance payments, security deposits, deposits (including deposits related to any Purchased Contract) and prepaid items exclusively related to the Product or the Business described on Section 1.1(i)(A) of the Seller Schedule. Seller’s good faith estimate of the amount and value of the Purchased Prepaid Items as of the Execution Date, calculated in accordance with US GAAP, is set forth on Section 1.1(i)(B) of the Seller Schedule.
“Purchased Regulatory Approvals” means all Regulatory Approvals held by the Seller or any of the Selling Affiliates exclusively related to the Product or the Business, as listed on Section 1.1(j) of the Seller Schedule, in each case in the form currently maintained by Seller or any of the Selling Affiliates (e.g., electronic).
“Purchaser Material Adverse Effect” means any event, fact, condition, occurrence, change or effect that prevents, materially impedes or materially delays or is reasonably likely to prevent, materially impede or materially delay the consummation by Purchaser of the transactions contemplated by this Agreement or the Ancillary Agreements.
“Purchaser Schedule” means the disclosure schedule of Purchaser delivered to the Seller in connection herewith.
“Purchaser Specified Representations” means the representations and warranties of Purchaser set forth in Section 4.1 (Organization), Section 4.2 (Authority; Execution and Delivery; Enforceability) and Section 4.7 (Brokers and Finders).
“Purchaser Tax Act” means any election under any provision of applicable Laws effective for the Pre-Closing Tax Period that is made after the Closing, and any other action taken after the Closing on the Closing Day, by Purchaser, any of its Affiliates or any of their transferees, that increases the amount of a liability for Taxes with respect to the Business or the Purchased Assets for any Pre-Closing Tax Period.
“Reference Exchange Rate” means, with respect to any day, the exchange rate (a) as published by Reuters (WMR Reuters fixing) referring to the exchange rate at 4:00 p.m. London Time on the relevant day or (b) if no rates are published on that day, on the latest day before that day for which such rates are published.
13
“Registered Intellectual Property” means Intellectual Property that is the subject of an application, certificate, filing, registration, or other document issued by, filed with, or recorded by, any Governmental Authority in any jurisdiction.
“Regulatory Approvals” means, with respect to the Product or any Subject Product, any and all written approvals, licenses, registrations (except manufacturing establishment registrations) or authorizations of any Governmental Authority necessary to commercially distribute, sell or market the Product or such Subject Product, including, where applicable, (a) pricing or reimbursement approvals, (b) pre- and post-approval marketing authorizations and (c) labeling approvals.
“Regulatory Documentation” means original documents or, to the extent original documents are not reasonably available, copies thereof, in any format, in the control of the Seller or any of the Selling Affiliates as of the Closing, evidencing all Purchased Regulatory Approvals, Product Specifications and correspondence with any Governmental Authority (including minutes and official contact reports relating to any communications with any Governmental Authority) and all other books and records relating to the development and regulatory approval of the Product, in each case, exclusively related to the Product or the Business (including research files, expert reports, research lab and engineering notes, regulatory applications, submissions, whether approved, pending, or withdrawn), formulation data, any other data (including clinical and pre-clinical data), submissions and filings in connection with the foregoing and relevant supporting documents including all regulatory drug lists, support for marketing and labeling claims and adverse event reports (periodic and expedited), in each case, exclusively related to the Product or the Business, but excluding any Seller Names and Marks, in each case to the extent in the control of Seller or any of its Affiliates and in each case, in the form currently maintained by Seller (e.g., electronic).
“Regulatory Laws” means all Laws applicable to the Exploitation of drugs or to the licensing, permitting, certification, accreditation, or registration of, and standards for, establishments involved in any such Exploitation, including: (i) the FDCA; (ii) the U.S. Public Health Service Act (42 U.S.C. § 201 et seq.) and all rules and regulations promulgated thereunder; (iii) GCP, GLP, and GMP; (iv) all terms, conditions, and requirements of any Regulatory Approvals; (v) any Laws pertaining to health care fraud and abuse, including the federal Anti-Kickback Statute (42 U.S.C. § 1320a-7b(b)), the civil False Claims Act (31 U.S.C. §§ 3729 et seq.), the federal sunshine and open payments law (42 U.S.C. § 1320a-7h), the federal health care program exclusion provisions (42 U.S.C. § 1320a-7), the Civil Monetary Penalties Act (42 U.S.C. § 1320a-7a), and all rules and regulations promulgated under any of the foregoing; (vi) Laws governing health care programs of any Governmental Authority and drug price reporting requirements; (vii) the federal Medicare and Medicaid statutes (Title XVIII and Title XIX of the Social Security Act) and all rules and regulations promulgated thereunder; (viii) the Patient Protection and Affordable Care Act, Pub. L. No. 111-148 (as amended); (ix) Laws pertaining to the licensing of, Permits for, and standards for drug manufacturers and distributors; and (x) any comparable international, European Union, national, foreign, state, or local Laws that address the subject matter of the foregoing.
“Sales and Promotional Materials” means advertisements, marketing and promotional materials, relating exclusively to the Product, but excluding any Seller Names and Marks and
14
any intellectual property and intellectual property rights of any third party that are contained or depicted therein.
“Seller Names and Marks” means (a) the OMEROS Trademark, (b) the PHARMACOSURGERY Trademark, (c) all other Trademarks to the extent used or held for use, in each case, as of the Closing Date, on any product packaging, product inserts or product labels of the Product (other than the Purchased Trademarks), and (d) all domain names and social media tags, handles and other identifiers consisting of or featuring any of the Trademarks referred to in clauses (a) or (b) above, except such domain names and social media tags that also contain or feature any of the Purchased Trademarks.
“Seller Products” means any and all products that are or will be Manufactured or Exploited by or on behalf of the Seller or any of its Affiliates other than the Subject Products.
“Seller Schedule” means the disclosure schedule of the Seller delivered to Purchaser in connection herewith.
“Seller Specified Representations” means the representations and warranties of the Seller set forth in Section 3.1 (Organization and Standing), Section 3.2 (Authority; Execution and Delivery; Enforceability), Section 3.4(a) (Title), Section 3.6 (Intellectual Property) and Section 3.13 (Brokers and Finders).
“Selling Affiliates” means all of the Affiliates of the Seller that own any Purchased Assets or that have Liabilities constituting any Assumed Liabilities, each of which is listed on Section 1.1(k) of the Seller Schedule.
“Shared Contract” has the meaning set forth in Section 2.5(c).
“Solvent” when used with respect to any Person, means that, as of any date of determination, (a) the amount of the “fair saleable value” of the assets of such Person will, as of such date, exceed (i) the sum of all stated “liabilities of such Person, including contingent and other liabilities,” as of such date, as such quoted terms are generally determined in accordance with applicable Laws governing determinations of the solvency of debtors, and (ii) the amount that will be required to pay the probable liabilities of such Person, as of such date, on its existing debts (including contingent and other liabilities) as such debts become absolute and mature, (b) such Person will not have, as of such date, an unreasonably small amount of capital for the operation of the businesses in which it is engaged or proposed to be engaged following such date, and (c) such Person will be able to pay its liabilities, including contingent and other liabilities, as they mature. For purposes of this definition, “not have an unreasonably small amount of capital for the operation of the businesses in which it is engaged or proposed to be engaged” and “able to pay its liabilities, including contingent and other liabilities, as they mature” means that such Person will be able to generate enough cash from operations, asset dispositions or refinancing, or a combination thereof, to meet its obligations as they become due.
“Straddle Tax Period” means any taxable period beginning on or prior to the Closing Date and ending after the Closing Date.
15
“Subject Products” means, collectively, (a) the Product, (b) any Combination Products and (c) any line extensions, synthetic versions, other administration forms, presentations, dosages, formulations, improvements or next generation products for or of the Product or any Combination Product, whether prescription or over-the-counter.
“(Sub)license Revenue” means, amounts received by Purchaser or any of its Affiliates in any form of consideration (including upfront payments, milestones, minimum royalty payments or sales-based revenues) for the grant of a (sub)license of rights to sell a Subject Product in any country other than the United States, but shall not include amounts received as a loan at prevailing market interest rates, amounts received for the purchase of an equity interest in licensee at fair market value or fees for services rendered on customary terms.
“Subsidiary” of any Person means another Person, an amount of the voting securities, other voting ownership or voting partnership interests of which is sufficient to elect at least a majority of its Board of Directors or other governing body (or, if there are no such voting interests, fifty percent (50%) or more of the equity interests of which) is owned directly or indirectly by such first person or by another Subsidiary of such first person.
“SVB Consent” means the Consent and Second Amendment to Loan and Security Agreement, dated as of December 1, 2021 by and between Silicon Valley Bank and Seller.
“Tax” or “Taxes” means all forms of taxation imposed by any U.S. Federal, state, provincial, local, non-U.S. or other Taxing Authority, including income, franchise, profits, gross receipts, capital gains, capital, capital stock, real and personal property, sales, use, goods and services, excise, unemployment, payroll, worker’s compensation, social security, employment, production, privilege, lease, service, license, service use, estimated, excise, value added, ad valorem, severance, stamp, transfer, documentary, registration, business and occupation, premium, escheat or unclaimed property, windfall profits, utility, environmental, communications, disability, alternative or add on minimum, recapture, withholding and other taxes, assessments, charges, duties, customs, fees, levies or other governmental charges of any kind, including any interest, penalties and additions thereto.
“Tax Proceeding” means any examination, audit, request for information, investigation, hearing, litigation, legal action, or administrative or judicial proceeding or contest relating to Taxes with a Governmental Authority.
“Tax Return” means any report, return, document, declaration or other information or filing required to be supplied to any Taxing Authority with respect to Taxes, including any amendment made with respect thereto.
“Taxing Authority” means any Governmental Authority responsible for the collection or administration of Taxes.
“Third Party” means any Person other than Purchaser, Seller and their respective Affiliates.
“Trademark” means any word, name, symbol, color, designation or device or any combination thereof, whether registered or unregistered, including any trademark, trade dress,
16
service mark, logo, slogan or other designation of origin and all registrations and applications relating thereto, as well as all goodwill associated with or symbolized thereby.
“Trademark Assignment Agreement” means that certain Trademark Assignment Agreement to be entered into as of the Closing Date among Purchaser and the Seller or the Selling Affiliate(s) party thereto, for the assignment to Purchaser of the Seller’s and the Selling Affiliates’ right, title and interest to and in the Registered Intellectual Property that are Trademarks, substantially in the form of Exhibit C attached hereto.
“Transfer Taxes” means all transfer, recording, ad valorem, privilege, documentary, gross receipts (other than Washington B&O Tax), registration, filing, conveyance, excise, license, stamp or similar Taxes (but excluding Indirect Taxes) imposed in respect of the transfer of the Business or the Purchased Assets pursuant to this Agreement.
“Transitional Services Agreement” means that certain Transitional Services Agreement to be entered into as of the Closing Date between Purchaser and the Seller, substantially in the form of Exhibit D attached hereto, with such mutually agreed supplements and modifications to the terms and conditions for the services described on Schedule 2.01 thereto as necessary to detail and operationalize the services (and related fees) described therein.
“Transitional Trademark License Agreement” means that certain Transitional Trademark License Agreement to be entered into as of the Closing Date between Purchaser and the Seller, substantially in the form of Exhibit E attached hereto.
“US GAAP” means generally accepted accounting principles in the United States.
“Valid Claim” means, with respect to a particular country, (a) any claim of an issued and unexpired Patent in such country that (i) has not been held permanently revoked, unenforceable or invalid by a decision of a court or Governmental Authority of competent jurisdiction, which decision is unappealable or unappealed (not including a petition for writ of certiorari to the U.S. Supreme Court or its foreign equivalent) within the time allowed for appeal and (ii) has not been abandoned, disclaimed, denied or admitted to be invalid or unenforceable in such country or (b) any claim of a Patent application that has been pending for seven years or less, which claim is being diligently prosecuted and has not been abandoned or finally disallowed without the possibility of appeal or re-filing of the application.
“Washington B&O Tax” means the business and occupation tax imposed by the State of Washington.
17
Index of Additional Defined Terms
Defined Term | Section |
| |
Accountant | Section 2.7(i)(ii) |
| |
Acquired Employee | Section 6.11(a) |
| |
Additional Payments | Section 2.7(c)(i) |
| |
Approvals | Section 2.5(a) |
| |
Assumed Liabilities | Section 2.3 |
| |
Audit Notice | Section 2.7(e) |
| |
Auditor | Section 2.7(e) |
| |
Base United States Royalty | Section 2.7(c)(i) |
| |
Basket | Section 9.4(a) |
| |
Claim Notice | Section 9.3(b)(i) |
| |
Closing | Section 2.6 |
| |
Closing Date | Section 2.6 |
| |
Closing Date Statement | Section 2.7(h)(ii) |
| |
Closing Inventory Value | Section 2.7(h)(ii) |
| |
Closing Prepaid Items Value | Section 2.7(h)(ii) |
| |
Debt Commitment Letter | Section 4.9(a) |
| |
Debt Financing | Section 4.9(a) |
| |
Debt Financing Parties | Section 11.15 |
| |
Defective Product | Section 6.17(c) |
| |
Definitive Agreements | Section 6.22(a)(ii) |
| |
Direct Claim | Section 9.3(b)(i) |
| |
Employment Guarantee Period | Section 6.11(c) |
| |
Enforceability Exceptions | Section 3.2(a) |
1
Estimated Closing Statement | Section 2.7(h)(i) |
| |
Estimated Inventory Value | Section 2.7(h)(i) |
| |
Estimated Prepaid Items Value | Section 2.7(h)(i) |
| |
Excluded Assets | Section 2.2 |
| |
Ex-United States Royalty | Section 2.7(c)(i) |
| |
Final Allocation | Section 10.1(a) |
| |
Financial Records | Section 2.7(d) |
| |
Foreign Merger Control Laws | Section 4.3(b) |
| |
Governance Documents Section 3.3(a) | |
| |
Inactive Employee Section 6.11(a) | |
| |
Indemnification Objection Notice | Section 9.3(b)(iii) |
| |
Indemnification Objection Period | Section 9.3(b)(iii) |
| |
Indemnified Party | Section 9.3(a) |
| |
Indemnifying Party | Section 9.3(a) |
| |
Inside Date | Section 2.6 |
| |
Judgment | Section 3.3(a) |
| |
Lenders | Section 4.9(a) |
| |
Losses | Section 9.1 |
| |
MCDA | Section 6.3(a) |
| |
Milestone Payment | Section 2.7(b) |
| |
Notice | Section 3.12(a) |
| |
Notice of Objection | Section 2.7(i)(i) |
| |
Objection Period | Section 2.7(i)(i) |
| |
Outside Date | Section 8.1(b)(i) |
| |
Owned Registered Product IP | Section 3.6(a) |
2
Parent Guarantor | Preamble |
| |
Pre-Closing Period | Section 6.1(a) |
| |
Privileged Information | Section 6.10(b) |
| |
Privileges | Section 6.10(b) |
| |
Purchase Price | Section 2.7(a) |
| |
Purchased Assets | Section 2.2 |
| |
Purchased Contracts | Section 2.1(a) |
| |
Purchaser Indemnitees | Section 9.1 |
| |
Royalty | Section 2.7(c)(i) |
| |
Royalty Term | Section 2.7 |
| |
Real Property Leases | Section 3.5 |
| |
Required Seller Assets | Section 6.16 |
| |
Required Terms | Section 6.11(a) |
| |
Resolution Period | Section 2.7(i)(ii) |
| |
Retained Liabilities | Section 2.4 |
| |
Retained Shared Contracts | Section 6.29 |
| |
Seller 401(k) Plan | Section 6.11(g) |
| |
Seller Indemnitees | Section 9.2 |
| |
Seller NDC Codes | Section 2.2(o) |
| |
Solvent | Section 4.6 |
| |
Sublicense Revenue Royalty | Section 2.7(c)(i) |
| |
Supplemental United States Royalty | Section 2.7(c)(i) |
| |
Supplemental United States Royalty Termination Event | Section 2.7 |
| |
Third Party Claim | Section 9.3(a) |
| |
Upfront Payment | Section 2.7(a) |
3
WARN Act | Section 3.10(d) |
ARTICLE II
Purchase and Sale
Section 2.1Purchased Assets. Subject to the terms and conditions set forth in this Agreement, at and effective as of the Closing, the Seller shall, and shall cause the Selling Affiliates to, sell, convey, assign, transfer and deliver to Purchaser, and Purchaser shall, or shall cause its Affiliates to, purchase and accept, all of the Seller’s and the Selling Affiliates’ rights, title and interest in and to the Purchased Assets held by the Seller or the Selling Affiliates as of the Closing Date, free and clear of all Liens, other than Permitted Liens. As used in this Agreement, “Purchased Assets” means the following rights and assets of the Seller and the Selling Affiliates:
(a)all rights under each Contract (i) set forth on Section 2.1(a) of the Seller Schedule, (ii) entered into by the Seller or any of its Affiliates between the Execution Date and the Closing Date in accordance with Section 6.1 that is exclusively related to the Product or the Business or (iii) that constitutes a Shared Contract, but only the portion of such Shared Contract exclusively related to the Business ((i)-(iii), collectively, “Purchased Contracts”);
(b)the Purchased Regulatory Approvals;
(c)the Purchased Intellectual Property;
(d)the Purchased Domain Names;
(e)the Purchased Know-how;
(f)the Purchased Inventory;
(g)the Purchased Equipment;
(h)the Purchased Prepaid Items;
(i)the Books and Records and Regulatory Documentation, but excluding in each case any attorney work product, attorney-client communications and other items protected by attorney-client or other legal privilege unless such books and records can be transferred without losing such privilege; provided, that to the extent there are books and records that include information related to the Product or the Business that do not constitute Books and Records, upon Purchaser’s reasonable written request, which request specifically identifies such books, records and/or information, Seller will provide copies thereof, appropriately redacted to exclude unrelated information;
(j)all goodwill associated with the Business;
(k)all guaranties, warranties, indemnities, rights of contribution, rights to refunds, rights of reimbursement and other rights of recovery and similar rights that have been
4
made by any predecessors in title, manufacturers or suppliers and other third parties relating to the Exploitation of the Purchased Assets from and after the Closing Date; and
(l)all claims, counterclaims, defenses, causes of action, demands, judgments, rights of recovery, rights of set-off, rights of subrogation and all other rights of any kind against any third party relating to the Purchased Assets.
Section 2.2Excluded Assets. The Purchased Assets shall not include, and there shall be excluded from the sale, conveyance, assignment, transfer or delivery to Purchaser hereunder, and each of the Seller and its Affiliates shall retain all of their existing right, title and interest in and to, any assets, properties, rights or interests other than those specifically listed or described in Section 2.1 (all such assets, properties, rights or interests not so listed or described, collectively, the “Excluded Assets”). For the avoidance of doubt, the Excluded Assets shall include, and the Purchased Assets shall not include:
(a)all Seller Products;
(b)all Accounts Receivable at any time due and owed to the Seller or any of its Affiliates, including Accounts Receivable arising from sales of any of the Product by or on behalf of the Seller or any of its Affiliates on or prior to the Closing Date;
(c)any losses, loss carryforwards and rights to receive refunds, credits and loss carryforwards with respect to any and all Taxes of the Seller or any of its Affiliates;
(d)any refund of Taxes if a liability for such Taxes would constitute a Retained Liability;
(e)all Tax Returns, Tax records, related workpapers and other similar Tax information of the Seller and its Affiliates;
(f)the books and records of the Seller and its Affiliates other than the Books and Records transferred pursuant to Section 2.1(i);
(g)any current and prior insurance policies and insurance Contracts, all rights of any nature with respect thereto, together with any claim, action or other right that the Seller or any of its Affiliates may have for insurance coverage under any such insurance policies or Contracts;
(h)any Intellectual Property or other intellectual property right that is not included in the Purchased Intellectual Property, the Purchased Domain Names or the Purchased Know-how, including the Seller Names and Marks;
(i)all Contracts (including the Excluded Contracts) of the Seller or any of its Affiliates other than the Purchased Contracts;
(j)any leased or owned real property;
5
(k)all IT equipment and all other tangible personal property of the Seller and its Affiliates other than the Purchased Inventory and the Purchased Equipment;
(l)any and all obligations, and all assets arising out of or related to Seller’s employee benefit plans, programs or arrangements (including the Business Benefit Plans) for the present or past employees, independent contractors, consultants, or agents of the Seller;
(m)all cash and cash equivalents (including marketable securities and short term investments), corporate credit cards, and deposits held by the Seller or any of its Affiliates, in each case, including those related to the Business;
(n)all guaranties, warranties, indemnities, rights of contribution, rights to refunds, rights of reimbursement and other rights of recovery and similar rights that have been made by any predecessors in title, manufacturers or suppliers and other third parties relating to the Excluded Assets;
(o)the ten or eleven digit three-segment numbers identified as a National Drug Code that is associated with any Product as assigned by FDA to Seller (“Seller NDC Codes”);
(p)all claims, counterclaims, defenses, causes of action, demands, judgments, rights of recovery, rights of set-off, rights of subrogation and all other rights of any kind against any third party, relating to any Retained Liabilities or the Excluded Assets; or
(q)all of the Seller’s and its Affiliates’ rights under this Agreement and the Ancillary Agreements and all books, documents, records, files or other items relating to the negotiation and consummation of this Agreement and the other transactions contemplated by this Agreement or the Ancillary Agreements or otherwise prepared in connection with the sale of the Purchased Assets, including all confidential communications with legal counsel representing the Seller or its Affiliates and the right to assert attorney-client privilege with respect thereto.
Section 2.3Assumed Liabilities. Subject to the terms and conditions set forth in this Agreement, at the Closing, the Seller shall and shall cause the Selling Affiliates, to assign to Purchaser and Purchaser shall assume, and agree to pay or otherwise perform or discharge when due, the Assumed Liabilities. As used in this Agreement, the term “Assumed Liabilities” means, except as expressly provided for otherwise in this Section 2.3 or Section 2.4, any and all Liabilities arising out of, in respect of or related to (i) the use, ownership, possession, conduct or operation of the Purchased Assets, (ii) the Exploitation of the Subject Products, or (iii) the conduct or operation of the Business, in each case, on or after the Closing Date, including:
(a)such Liabilities of the Seller or any of its Affiliates under any Purchased Contract;
(b)such Liabilities of Purchaser under any Shared Contract, in accordance with Section 2.5(c);
6
(c)such Liabilities of the Seller or any of its Affiliates with respect to the Purchased Regulatory Approvals or incurred to obtain Regulatory Approvals or any other permits in any jurisdiction;
(d)such Liabilities arising out of or relating to, including all Liabilities arising out of or relating to any Action in respect of, any recalls, product liability, Intellectual Property or other intellectual property infringement, breach of warranty or similar claim for injury to person or property, in each case to the extent arising out of, in respect of or related to any Subject Product sold by Purchaser, its Affiliates or (sub)licensees on or after the Closing Date;
(e)such Liabilities arising under any Environmental Laws;
(f)Assumed Tax Liabilities;
(g)all Liabilities associated with the employment of the Acquired Employees by Purchaser or its Affiliates with respect to any period on or after the Closing Date;
(h)all Liabilities for chargebacks, rebates, cash discounts and fees for services paid to wholesalers and distributors or to ASC and hospital customers and other comparable gross to net adjustments, including Liabilities for discounts and rebates payable under U.S. drug price reporting and discounting programs such as the Medicaid Drug Rebate Program, 42 U.S.C. § 1396r-8, Medicare Part B Average Sales Price reporting, 42 U.S.C. § 1395w–3a, the 340B Program, 42 U.S.C. § 256b, and the Department of Veterans Affairs Drug Price reporting program, 38 U.S.C. § 8126, including with respect to discounts and rebates due to any recalculations or restatements of metrics reported under such programs, in each case to the extent relating to or arising from the sale of the Subject Products sold on or after the Closing Date;
(i)all trade accounts, accrued receipts and accounts payable of the Purchaser and its Affiliates arising out of the operation or conduct of the Business on or after to the Closing; and
(j)all Liabilities for which the Purchaser or any of its Affiliates expressly has responsibility pursuant to this Agreement or the Ancillary Agreements.
Section 2.4Retained Liabilities. Except for the Assumed Liabilities, Purchaser shall not assume, nor become responsible for, and Seller and the Selling Affiliates shall retain and be responsible for and shall pay, perform and discharge when due the Retained Liabilities. As used in this Agreement, the term “Retained Liabilities” means any and all Liabilities arising out of, in respect of or related to (i) the use, ownership, possession, conduct or operation of the Purchased Assets, (ii) the Exploitation of the Product or (iii) or the conduct or operation of the Business, in each case, prior to the Closing Date, including:
(a)such Liabilities of the Seller or any of its Affiliates under any Purchased Contract;
(b)such Liabilities of the Seller or any of its Affiliates under any Shared Contract, in accordance with Section 2.5(c).
7
(c)such Liabilities of the Seller or any of its Affiliates with respect to the Purchased Regulatory Approvals or incurred to obtain Regulatory Approvals or any other permits in any jurisdiction;
(d)such Liabilities arising out of or relating to, including all Liabilities arising out of or relating to any Action in respect of, any recalls, product liability, Intellectual Property or other intellectual property infringement, breach of warranty or similar claim for injury to person or property, in each case, to the extent arising out of, in respect of or related to any Product sold by Seller, its Affiliates or (sub)licensees prior to the Closing Date;
(e)such Liabilities arising under any Environmental Laws;
(f)all Liabilities for Taxes of the Seller and its Affiliates other than Assumed Tax Liabilities;
(g)all Liabilities associated with (i) the employment or termination of the Acquired Employees by the Seller or its Affiliates with respect to any period on or prior to the Closing Date and (ii) employees, independent contractors, consultants, or agents of the Seller, other than the Acquired Employees, with respect to any period; and
(h)all Liabilities for chargebacks, rebates, cash discounts and fees for services paid to wholesalers and distributors or to ASC and hospital customers and other comparable gross to net adjustments, including Liabilities for discounts and rebates payable under U.S. drug price reporting and discounting programs such as the Medicaid Drug Rebate Program, 42 U.S.C. § 1396r-8, Medicare Part B Average Sales Price reporting, 42 U.S.C. § 1395w–3a, the 340B Program, 42 U.S.C. § 256b, and the Department of Veterans Affairs Drug Price reporting program, 38 U.S.C. § 8126, including with respect to discounts and rebates due to any recalculations or restatements of metrics reported under such programs, in each case to the extent relating to or arising from the sale of the Product sold prior to the Closing Date;
(i)all trade accounts, accrued receipts and accounts payable of the Seller and its Affiliates arising out of the operation or conduct of the Business prior to the Closing;
(j)all Liabilities to the extent arising out of or related to the Excluded Assets; and
(k)all Liabilities for which the Seller or any of the Selling Affiliates expressly has responsibility pursuant to this Agreement or the Ancillary Agreements.
Section 2.5Non-Assignment; Consents.
(a)Notwithstanding anything in this Agreement to the contrary, this Agreement shall not constitute an agreement to sell, assign, transfer or convey any Purchased Asset (or any Assumed Liabilities thereunder) if an attempted sale, assignment, transfer or conveyance thereof would be prohibited by Law or would, without the approval, authorization or consent of, filing with, notification to, or granting or issuance of any license, order, waiver or permit by, any Governmental Authority or any third party counterparty to a Purchased Contract or a Shared Contract (collectively, “Approvals”), (i) constitute a breach thereof or contravention
8
thereof, (ii) be ineffective, void or voidable, or (iii) adversely affect the rights thereunder of the Seller, Purchaser, or any of their respective officers, directors, agents or Affiliates, unless and until such Approval is obtained.
(b)The Seller and Purchaser shall use commercially reasonable efforts to obtain, or cause to be obtained, any Approval (other than the Approvals governed by Section 6.4) required to sell, assign or transfer any Purchased Asset and transfer the Assumed Liabilities from and after the Closing. If any such Approval is not obtained prior to Closing, until the earlier of such time as such Approval is obtained or one year following the Closing Date, the Seller will use commercially reasonable efforts to cooperate with Purchaser to establish an agency type or any other similar arrangement reasonably acceptable to Purchaser and Seller intended to both (i) provide Purchaser, to the fullest extent practicable, the claims, rights and benefits of any such Purchased Assets, and (ii) cause Purchaser to bear all costs and Liabilities thereunder from and after the Closing in accordance with this Agreement (including by means of any subcontracting, sublicensing or subleasing arrangement). In furtherance of the foregoing, Purchaser will promptly pay, perform or discharge when due any Assumed Liability arising thereunder after the Closing Date and the Seller shall, and shall cause its Affiliates to, without further consideration therefor, promptly pay and remit to Purchaser all monies, rights and other consideration received thereunder. Notwithstanding anything in this Agreement to the contrary, no Party or any of its Affiliates shall be required to pay compensation to any third party, commence any Action or offer or grant any accommodation (financial or otherwise, including any accommodation or arrangement to remain secondarily liable or contingently liable for any Assumed Liability) to any third party to obtain any such third party consent or other Approval. Purchaser agrees, subject to applicable Law and any overriding obligations of confidentiality, to provide any such evidence as to financial capability, resources and creditworthiness of Purchaser or its Affiliates as may be reasonably requested by any third party whose consent or approval is sought in connection with the transactions contemplated by this Agreement. For the avoidance of doubt, (1) no representation, warranty or covenant of the Seller or any Selling Affiliate contained in this Agreement and the Ancillary Agreements shall be breached or deemed breached, and no condition shall be deemed not satisfied, based solely on the failure to obtain any such Approvals pursuant to this Section 2.5(b) or (c) and (2) no covenant of the Seller or any Selling Affiliate contained in this Agreement and the Ancillary Agreements shall be breached or deemed breached, and no condition shall be deemed not satisfied, based solely on any Action commenced or threatened by or on behalf of any Person arising out of or relating to the failure to obtain any such Approvals pursuant to this Section 2.5(b) or (c).
(c)Any Contract to be assigned, transferred and conveyed in accordance with Section 2.1 and set forth on Section 2.5(c) of the Seller Schedule that does not exclusively relate to the Product or the Business (each, a “Shared Contract”) shall be assigned, transferred and conveyed only with respect to (and preserving the meaning of) those parts that exclusively relate to the Product or the Business, to Purchaser, if so assignable, transferrable or conveyable, or appropriately amended prior to, on or after the Closing, so that (i) Purchaser shall be entitled to the rights and benefits of those parts of the Shared Contract that exclusively relate to the Product or the Business and shall assume only the related portion of any Liabilities thereunder to the extent such Liabilities are Assumed Liabilities and (ii) the Seller shall be entitled to the rights and benefit of those parts of the Shared Contract that do not relate exclusively to the Product or the Business and shall only retain Liabilities thereunder to the extent such Liabilities are
9
Retained Liabilities; provided, that (1) in no event shall any Person be required to assign (or amend), either in its entirety or in part, any Shared Contract that is not assignable (or cannot be amended) by its terms without obtaining one or more Approvals and (2) if any Shared Contract cannot be so partially assigned by its terms, or cannot be amended, without such Approval or Approvals, until the earlier of such time as such Approval or Approvals are obtained and one year following the Closing Date, then the Seller and Purchaser will use their respective commercially reasonable efforts to cooperate to establish an agency type or other similar arrangement reasonably satisfactory to the Seller and Purchaser intended to (A) provide Purchaser, to the fullest extent practicable under such Shared Contract, the claims, rights and benefits of the parts that relate exclusively to the Product or the Business from and after the Closing, (B) cause Purchaser to promptly pay, perform or discharge when due all Assumed Liabilities thereunder from and after the Closing in accordance with this Agreement, (C) provide the Seller, to the fullest extent practicable under such Shared Contract, the claims, rights and benefits of those parts that do not relate exclusively to the Product or the Business from and after the Closing and (D) cause Seller to promptly pay, perform or discharge when due all Retained Liabilities thereunder from and after the Closing in accordance with this Agreement (including by means of any subcontracting, sublicensing or subleasing arrangement). Notwithstanding anything in this Agreement to the contrary, no Party shall be required to pay compensation to any third party, commence or participate in any Action or offer or grant any accommodation (financial or otherwise, including any accommodation or arrangement to remain secondarily liable or contingently liable for any Assumed Liability) to any third party in connection with its obligations under this Section 2.5(c).
(d)For so long as the Seller or any Selling Affiliate holds any Purchased Assets or is a party to any Shared Contracts after the Closing and provides Purchaser any claims, rights and benefits of any such Purchased Asset or Shared Contracts (in the case of such Shared Contracts, to the extent related to the Business) pursuant to an arrangement described in Section 2.5(b) or (c), Purchaser shall indemnify and hold the Seller and its Affiliates harmless from and against all Losses incurred or asserted as a result of the Seller’s or its Affiliates’ post-Closing direct or indirect ownership, management or operation of any such Purchased Assets or Shared Contracts (in each case, in accordance with this Agreement), except to the extent resulting from the Seller’s or any of its Affiliates’ Fraud, gross negligence or willful misconduct. Notwithstanding anything contained herein to the contrary, any transfer or assignment to Purchaser of any Purchased Asset or any part of a Shared Contract that shall require an Approval as described above in this Section 2.5 shall be made subject to such Approval being obtained; provided, that upon receipt of such Approval, such transfer and assignment shall automatically and without further action be effected in accordance with the terms of this Agreement.
Section 2.6The Closing. Unless otherwise agreed by the Parties, the closing of the Acquisition (the “Closing”) shall take place at the offices of Covington & Burling LLP, 415 Mission Street, San Francisco, California or by the exchange of remote signatures, on the sixth Business Day following the date on which the last condition to the obligations of the Parties to consummate the Acquisition (other than those conditions to be satisfied at the Closing, but subject to satisfaction or waiver of such conditions at the Closing) has been satisfied (or, to the extent permitted by applicable Law, waived by the party entitled to waive such condition); provided, that in no event shall the Closing take place before the 30th calendar day following the date hereof (the “Inside Date”) without the prior written consent of Purchaser (the actual date of
10
the Closing, the “Closing Date”). The Closing shall be deemed to have occurred at 12:01 a.m., Eastern time, on the Closing Date, such that Purchaser shall be deemed the owner of the Purchased Assets on and after the Closing Date.
Section 2.7Consideration.
(a)Upon the terms and subject to the conditions set forth in this Agreement, in consideration for the sale and purchase of the Purchased Assets and the other transactions contemplated hereby, Purchaser shall: (i) on the Closing Date, pay to the Seller an upfront cash payment equal to the sum of (A) $125,000,000, (B) minus 50% of the filing fees paid by or on behalf of Purchaser under the HSR Act prior to the Closing Date, (C) plus the amount, if any, by which the Estimated Inventory Value exceeds the Inventory Value Target Amount, (D) minus the amount, if any, by which the Estimated Inventory Value is less than the Inventory Value Target Amount and (E) plus the Estimated Prepaid Items Value (collectively, the “Upfront Payment”); and (ii) pay to the Seller the Milestone Payment and Royalties, in each case to the extent payable under Section 2.7(b) and Section 2.7(c), respectively (the Milestone Payment and Royalties, as and when payable, together with the Upfront Payment (as adjusted in accordance with Section 2.7(h)-(k), the “Purchase Price”).
(b)Milestone Event; Milestone Payment. Within 30 days following a Milestone Event, Purchaser shall make a one-time lump sum payment to the Seller of $200,000,000 (the “Milestone Payment”). Notwithstanding the foregoing, if a Milestone Event occurs after the Execution Date and at least six Business Days prior to the Closing Date, then the Milestone Payment shall be payable on the Closing Date together with the Upfront Payment in accordance with Section 2.7(a) otherwise such payment shall be made in accordance with this Section 2.7(b).
(c)Royalties.
(i)Royalty Rates. During the Royalty Term, Purchaser shall pay to the Seller (A) with respect to each calendar month, a non-refundable, non-creditable royalty on the Net Revenue of the Subject Product(s) in the applicable territories at the royalty rates set forth below, with each payment due no later than the end of the second calendar month after such calendar month, provided, that with respect to calendar months ending January 31, 2022, February 28, 2022 and March 31, 2022, payment shall be due no later than May 31, 2022 and (B) with respect to each calendar quarter, a non-refundable, non-creditable royalty on the (Sub)license Revenue of the Subject Product(s) in the applicable territories at the royalty rate set forth below, with each payment due within 50 days after the end of the applicable calendar quarter (each, ((A) or (B)), the “Royalty,” and, together with the Milestone Payment, the “Additional Payments”). For example, royalties on Net Revenue pursuant to clause (A) above in respect of June 30, 2022 shall be due no later than August 31, 2022.
| Royalty Categories | Royalty Rate | Territory |
1. | Base United States Royalty | 30% Of Net Revenue | United States |
2. | Supplemental United States | 20% | United States |
11
| Royalty | Of Net Revenue | |
3. | Ex-United States Royalty | 15% Of Net Revenue | All countries other than the United States |
4. | (Sub)license Revenue Royalty | 15% Of (Sub)license Revenue | All countries other than the United States |
For clarity, if Purchaser or any Affiliate grants a license or (sub)license of rights to sell a Subject Product in any country other than the United States, royalties in respect of such license or (sub)license will be payable under Item 4 above on (Sub)license Revenue and no royalty under Item 3 shall be payable on Net Revenue by such licensee or (sub)licensee.
(ii)Royalty Reductions. Notwithstanding Section 2.7(c)(i):
(A)During the Royalty Term, following the first sale of a Generic Version in the United States, if annualized Net Revenues of Subject Products in the United States during any calendar quarter are at least [***] percent ([***]%) less than Net Revenues of Subject Products in the United States during the 12 calendar month period immediately preceding the date of such first sale of a Generic Version in the United States (“Generic Entry”), the Base United States Royalty payable under Section 2.7(c)(i) shall be reduced from thirty percent (30%) to [***] ([***]%) of Net Revenue in the next subsequent calendar month and in all future calendar months to the extent such Generic Version is continuing to be sold in the United States.
(B)(1) The Base United States Royalty shall be reduced to ten percent (10%) of Net Revenue and (2) the Supplemental United States Royalty shall be reduced to zero percent (0%), in each case with respect to Subject Products sold in the United States during any year (or any portion thereof) during which no Subject Product is eligible for Separate Payment.
(C)For purposes of this Section 2.7:
(1) | [***] |
(2) | “Royalty Term” means (1) with respect to the Base United States Royalty, on a Subject Product-by-Subject Product Basis, the period beginning on the Closing Date and ending on the expiration or |
* [***] CERTAIN IDENTIFIED INFORMATION HAS BEEN EXCLUDED FROM THE EXHIBIT BECAUSE IT BOTH (A) IS NOT MATERIAL AND (B) WOULD BE COMPETITIVELY HARMFUL IF PUBLICLY DISCLOSED
12
termination of the last Valid Claim of any patent included in the Purchased Intellectual Property covering such Subject Product in the United States, (2) with respect to the Supplemental United States Royalty, the period beginning on the Closing Date and ending on the earlier of (x) December 31, 2024 (inclusive) and (y) the occurrence of the Milestone Event and (3) with respect to the Ex-United States Royalty and the (Sub)license Revenue Royalty, on a Subject Product-by-Subject Product and country-by-country basis with respect to each applicable country outside the United States, the period beginning on the Closing Date and ending on the expiration or termination of the last Valid Claim of a transferred patent included in the Purchased Intellectual Property covering such Subject Product in such country.
(3) | “Separate Payment” means that the Centers for Medicare and Medicaid Services (“CMS”) pays for any Subject Product in addition to the packaged payment rate established by CMS for the applicable surgical procedure when used for the treatment of Medicare Part B beneficiaries in the ambulatory surgical center setting, or any substantially equivalent reimbursement status. |
(D)Royalty Reports. Each monthly or quarterly Royalty payment (as applicable) paid to the Seller shall be accompanied by a statement substantially in the form of Exhibit G prepared by Purchaser with respect to the applicable calendar month/quarter, setting forth a calculation of Net Revenue showing with reasonable specificity the gross revenues, the itemized deductions from gross revenues provided for in the definition of Net Revenue during such calendar month/quarter, any reduction taken under Section 2.7(c)(ii), any currency conversion taken under Section 2.7(f), a calculation of (Sub)license Revenue, if any, and a calculation of the Royalty due with respect to such Net Revenue or (Sub)license Revenue, as applicable, for such calendar month/quarter, in all cases, reported in United States Dollars, and any material updates regarding the sales and marketing plan.
(E)Annual Commercial Report. Each royalty report delivered with respect to December of a given year shall be accompanied by (i) a summary of field sales representative FTEs employed by Purchaser or its Affiliates or licensees in connection with promoting Subject Products during such calendar year and (ii) a copy of the then-current sales and marketing annual plan and provide a summary update on commercialization, sales and marketing activities relating to the Subject Products for the following calendar year. Purchaser’s initial sales and marketing plan shall be provided to the Seller within 30 days following Closing.
13
(iii)Right of Set-off. Purchaser may deduct and set off against any Royalty or Milestone Payment owed to the Seller under this Section 2.7(c) any Losses subject to indemnification under Section 9.1 (Indemnification by Seller). Neither Party or its Affiliates shall otherwise be entitled to deduct from, set off, holdback or otherwise reduce in any manner whatsoever any amounts owed to the other Party or any of its Affiliates.
(d)Financial Records. Purchaser shall, and shall cause its controlled Affiliates and (sub)licensees to, keep or cause to be kept complete and accurate books and records pertaining to Net Revenue and the applicable royalty calculations in sufficient detail to calculate all amounts payable hereunder (“Financial Records”). Such books and records shall be retained until the later of (i) three years after the end of the period to which such books and records pertain and (ii) the expiration of the applicable tax statute of limitations (or any extensions thereof) or for such longer period as may be required by applicable Law.
(e)Audit. Not more often than once every calendar year, the Seller shall have the right to have an independent certified public accountant of nationally recognized standing, selected by the Seller and reasonably acceptable to Purchaser (the “Auditor”), perform a review of Purchaser’s Financial Records kept pursuant to Section 2.7(d) for the sole purpose of verifying the accuracy of the financial statements and reports related to calculation of any Additional Payment, with not less than 30 days’ advance written (the “Audit Notice”). Any such audit shall be conducted during regular business hours at the premises where such books and records are maintained. No later than the expiration of such 30-day period, Purchaser shall provide the Auditor with reasonable access to Financial Records for the purposes of such review. The Seller shall be responsible for the entire cost of any such audit, unless an audit reveals an underpayment by Purchaser of an amount greater than either (i) $500,000 or (ii) 5% or more of the total amount owing with respect to the audited period, in which case Purchaser shall bear the entire cost of any such audit. In the event an audit reveals an underpayment by Purchaser, Purchaser shall promptly pay the amount of such underpayment to the Seller, together with any applicable late payment interest owed with respect to such amount pursuant to Section 2.9(b). In the event an audit reveals an overpayment by Purchaser, Purchaser shall be allowed to credit the amount of such overpayment against future amounts.
(f)Currency Conversion. All payments to be made under this Agreement shall be made in United States Dollars. Net Revenue, (Sub)license Revenue and Royalties will be converted into United States Dollars using the applicable daily Reference Exchange Rates for the last day of the month in which such Net Revenue accrued.
(g)Efforts.
(i)On a country by country basis in the United States and European Major Markets, from the Closing Date and until the expiration of the Royalty Term in such country, Purchaser shall and shall cause any of its applicable Affiliates or its or their (sub)licensees to use Diligent Efforts to *[***].
(ii)From the Closing Date, for so long as Separate Payment is in effect or until Generic Entry has occurred, Purchaser, its Affiliates and (sub)licenses in the aggregate shall [***].
(iii)Purchaser shall not, and shall cause any of its applicable Affiliates or its or their (sub)licensees not to, take any action or omission [***].
* [***] CERTAIN IDENTIFIED INFORMATION HAS BEEN EXCLUDED FROM THE EXHIBIT BECAUSE IT BOTH (A) IS NOT MATERIAL AND (B) WOULD BE COMPETITIVELY HARMFUL IF PUBLICLY DISCLOSED
14
(h)Post-Closing Value Adjustment.
(i)Estimated Closing Statement. At least three Business Days prior to the Closing Date, Seller shall cause to be prepared and delivered to Purchaser a statement (“Estimated Closing Statement”) calculating in reasonable detail (A) Seller’s good faith estimate of the amount and value of the Purchased Inventory as of the Calculation Time calculated in accordance with US GAAP (the “Estimated Inventory Value”) and (B) Seller’s good faith estimate of the amount and value of the Purchased Prepaid Items as of the Calculation Time calculated in accordance with US GAAP (the “Estimated Prepaid Items Value”), in each case, together with reasonable supporting documentation relating thereto. Purchaser shall have the opportunity to review and comment on the Estimated Closing Statement and the Seller shall consider Purchaser’s comments in good faith; provided, that Purchaser’s review of and comments on the Estimated Closing Statement shall not operate to delay the Closing.
(ii)Closing Date Statement. Within 60 days after the Closing Date, Purchaser shall cause to be prepared and delivered to Seller a statement (the “Closing Date Statement”) calculating in reasonable detail (A) the actual amount and value of the Purchased Inventory as of the Calculation Time calculated in accordance with US GAAP (the “Closing Inventory Value”) and (B) the actual amount and value of the Purchased Prepaid Items as of the Closing Date calculated in accordance with US GAAP (the “Closing Prepaid Items Value”), in each case, together with reasonable supporting documentation relating thereto.
(i)Objections; Resolutions of Disputes.
(i)Unless Seller notifies Purchaser in writing within 30 days after Purchaser’s delivery of the Closing Date Statement (such 30-day period, the “Objection Period”) of any good faith objection to the computation of the Closing Inventory Value or Closing Prepaid Items Value, respectively, set forth therein (a “Notice of Objection”), the Closing Date Statement shall be final and binding. Following the delivery of the Closing Date Statement and solely for purposes of Seller’s review of the Closing Date Statement and preparation of any Notice of Objection, Purchaser shall permit Seller and its representatives to review the work papers of Purchaser relating to the Closing Date Statement. Any Notice of Objection shall specify in reasonable detail each item that Seller disputes, the amount in dispute for each such dispute and a description in reasonable detail of the basis for the objections set forth therein. Seller and Purchaser acknowledge that the sole purpose of the determination of the Closing Inventory Value and the Closing Prepaid Items Value is to adjust the Upfront Payment so as to reflect the difference between (A) the Closing Inventory Value and the Estimated Inventory Value and (B) the Closing Prepaid Items Value and the Estimated Prepaid Items Value, and that in order to do so the Closing Inventory Value and the Estimated Inventory Value as well as the
15
Closing Prepaid Items Value and the Estimated Prepaid Items Value need to be calculated in accordance with US GAAP as of the Calculation Time.
(ii)If Seller provides a Notice of Objection to Purchaser within the Objection Period, Seller and Purchaser shall, during the 30-day period following Purchaser’s receipt of the Notice of Objection (such 30-day period, the “Resolution Period”), attempt in good faith to resolve Seller’s objections. During the Resolution Period, Purchaser and its representatives shall, in accordance with Section 2.7(l), be permitted to review the work papers of Seller and its accountants relating to the Notice of Objection and the basis therefor. If Seller and Purchaser reach an agreement with respect to any of Seller’s objections, such agreement shall be reduced to writing and shall be final and binding on the Parties. If Seller and Purchaser are unable to resolve all such objections within the Resolution Period, the matters remaining in dispute shall be submitted to an independent certified public accountant of nationally recognized standing, mutually agreed by the Seller and Purchaser (the “Accountant”). The Accountant shall be engaged pursuant to an engagement letter among Seller, Purchaser and the Accountant on terms and conditions consistent with this Section 2.7(i). The Accountant shall be instructed, pursuant to such engagement letter, to resolve only those matters set forth in the Notice of Objection remaining in dispute and not to otherwise investigate any matter independently. Seller and Purchaser each agree to furnish to the Accountant access to such individuals and such information, books and records as may be reasonably required by the Accountant to make its final determination (any such information, books and records shall be provided to the other Party prior to its submission or presentation to the Accountant). Seller and Purchaser shall also instruct the Accountant to render its reasoned written decision as promptly as practicable but in no event later than 30 days from the date that the unresolved objections are submitted to the Accountant for review. With respect to each disputed line item, such decision, if not in accordance with the position of either Seller or Purchaser, shall not be in excess of the higher, nor less than the lower, of the amounts advocated by Purchaser in the Closing Date Statement, or Seller in the Notice of Objection with respect to such disputed line item. Except as Seller and Purchaser may otherwise agree, all communications between Seller and Purchaser or any of their respective representatives, on the one hand, and the Accountant, on the other hand, shall be in writing with copies simultaneously delivered to the non-communicating Party. The resolution of disputed items by the Accountant shall be final and binding on the Parties (absent Fraud or manifest error) and the determination of the Accountant shall constitute an arbitral award that is final, binding and non-appealable (absent Fraud or manifest error) and upon which a Judgment may be entered by a court having jurisdiction thereover. Each party will bear its own costs and expenses in connection with the resolution of such dispute by the Accountant. All fees and expenses of the Accountant in respect of services pursuant to this Section 2.7(i) shall initially be borne fifty percent (50%) by Seller and fifty percent (50%) by Purchaser; provided, that such fees and expenses of the Accountant shall be allocated between the Parties based upon the percentage which the portion of the contested amount not awarded to each Party bears to the amount actually contested by such Party in the Closing Date Statement or the Notice of Objection, as applicable, and will be determined by the Accountant in a manner consistent with such principles concurrently with and as part of its resolution. For example, if Purchaser claims an amount of $1,000, and if the Seller contests only $500 of the amount claimed by Purchaser, and if the Accountant ultimately resolves the dispute by awarding the Purchaser $300 of the $500 contested, then the fees and expenses of the Accountant will be allocated sixty percent (60%) (i.e., 300/500) to the Seller and forty percent (40%) (i.e., 200/500) to the Purchaser.
16
(j)Inventory Adjustment Payment. Promptly (but in any event within 10 Business Days) after the date on which the Closing Date Statement (and the Closing Inventory Value contained therein) becomes final and binding on Seller and Purchaser in accordance with Section 2.7(i), if (A) the Closing Inventory Value exceeds the Estimated Inventory Value, the Upfront Payment shall be adjusted upward by the amount of such excess, and Purchaser shall pay the amount of such excess to Seller and (B) if the Closing Inventory Value is less than the Estimated Inventory Value, the Upfront Payment shall be adjusted downward by the amount of such difference and Seller shall pay the amount of such difference to Purchaser.
(k)Prepaid Items Adjustment Payment. Promptly (but in any event within 10 Business Days) after the date on which the Closing Date Statement (and the Closing Prepaid Items Value contained therein) becomes final and binding on Seller and Purchaser in accordance with Section 2.7(i), (i) if the Closing Prepaid Items Value is less than the Estimated Prepaid Items Value, the Upfront Payment shall be adjusted downward by the amount of such difference, and Seller shall pay the amount of such difference to Purchaser, and (ii) if the Closing Prepaid Items Value is more than the Estimated Prepaid Items Value, the Upfront Payment shall be adjusted upward by the amount of such excess, and Purchaser shall pay the amount of such excess to Seller.
(l)Access to Seller’s Books and Records. Following the Closing and until the date the Closing Date Statement becomes final and binding pursuant to Section 2.7(i), Seller shall (i) provide or cause to be provided to Purchaser and its representatives access upon reasonable notice during normal business hours to the properties, books, Contracts, personnel and records of Seller, and Seller’s and its accountants’ work papers relevant to the preparation of the Closing Date Statement and the adjustments contemplated by this Section 2.7, (ii) provide Purchaser, upon Purchaser’s request, with copies of any such books, Contracts, records and work papers and (iii) cause its personnel to cooperate with Purchaser and respond to Purchaser’s requests for information promptly with respect thereto. The accountants of Seller shall not be obligated to make any work papers available to any Person under this Section 2.7 unless and until such Person has signed a customary confidentiality and hold harmless agreement relating to such access to work papers in form and substance reasonably acceptable to such accountants.
Section 2.8Closing Deliverables. At or prior to the Closing:
(a)The Seller and the Selling Affiliates, as applicable, shall deliver or cause to be delivered to Purchaser:
(i)the Purchased Assets; provided, that (1) with respect to tangible Purchased Assets, Books and Records, Regulatory Documentation, the Purchased Know-how and all other Purchased Assets described therein, delivery shall, unless the Parties otherwise mutually agree, be at the locations and on the timeframes to be mutually agreed at least three Business Days prior to the Closing and (2) the Seller may retain copies of the Purchased Contracts, Purchased Regulatory Approvals, Marketing Records, Regulatory Documentation and Purchased Know-how included within the Purchased Assets (and, prior to delivering or making available any files, documents, instruments, papers, books and records containing or comprising any Purchased Assets and prior to delivering or making any of the foregoing available to Purchaser, the Seller shall be entitled to redact from such files, documents, instruments, papers,
17
books and records any information to the extent that it does not relate to the Business or the Purchased Assets); provided, that any Confidential Information so retained shall remain subject to the applicable terms of the MCDA in accordance with the terms thereof and of Section 6.5;
(ii)a counterpart of the Domain Name Assignment Agreement, duly executed by each of the Seller or the applicable Selling Affiliate;
(iii)a counterpart of the Patent Assignment Agreement, duly executed by each of the applicable Selling Affiliates and, if applicable, the Seller;
(iv)a counterpart of the Trademark Assignment Agreement, duly executed by each of the applicable Selling Affiliates and, if applicable, the Seller;
(v)a counterpart of the Transitional Services Agreement, duly executed by the Seller;
(vi)a counterpart of the Transitional Trademark License Agreement, duly executed by the Seller;
(vii)a counterpart of the Assignment and Assumption Agreement, duly executed by the Seller;
(viii)a certificate duly executed by an authorized officer of the Seller stating that each of the conditions set forth in Section 7.2(a) and Section 7.2(b) has been satisfied;
(ix)offer letters, Purchaser’s standard form of Intellectual Property Agreement and other reasonable and customary employment documentation, , duly executed by 75% of the Business Employees who receive offer letters with the Required Terms pursuant to Section 6.11(a);
(x)a duly completed and executed IRS Form W-9 of Seller;
(xi)a duly completed and executed IRS Form W-8BEN-E of Omeros Ireland Limited, claiming the benefits of any relevant income tax treaty between its country of tax residence and the United States, including any article of such treaty relating to royalties or similar payments;
(xii)a certificate of the Secretary of State of the State of Washington as to the good standing of Seller as of a date not more than five Business Days prior to the Closing Date; and
(xiii)evidence of the consents from the Persons set forth in Section 2.8(a)(xiii) of the Seller Schedule, in each case, in a form reasonably satisfactory to Purchaser.
(b)Purchaser shall:
18
(i)make payment to one or more accounts designated in writing by the Seller (such designation to be made at least five Business Days prior to the Closing Date), in an amount equal to the Upfront Payment;
(ii)deliver to the Seller counterparts of the Domain Name Assignment Agreement, the Patent Assignment Agreement, the Trademark Assignment Agreement, the Transitional Services Agreement, the Transitional Trademark License Agreement, the Assignment and Assumption Agreement, each duly executed by Purchaser; and
(iii)deliver to the Seller a certificate duly executed by an authorized officer of Purchaser stating that each of the conditions set forth in Section 7.3(a) and Section 7.3(b) has been satisfied.
Section 2.9Payment Mechanics.
(a)All payments required to be made pursuant to the terms of this Agreement, including the payment of any portion of the Purchase Price (including Additional Payments) pursuant to Section 2.7 shall be made by wire transfer of immediately available funds in United States Dollars to such account as is designated in writing by the Seller to Purchaser (such designation to be made at least five Business Days prior to the Closing Date).
(b)The amount of any payments required to be made pursuant to the terms of this Agreement that is not paid when due shall be subject to late payment interest, to the extent not prohibited by Law, at a per annum rate equal to the U.S. Prime Rate, as reported in The Wall Street Journal, Eastern Edition, for the first date on which such payment was delinquent, plus two percent (2%), beginning on the first date on which such payment was delinquent and ending on the date on which such payment is made, calculated based on the actual number of days such payment is overdue.
Section 2.10Withholding Taxes. The Purchase Price and any other payment hereunder by Purchaser (or its Affiliates or any of its paying agents) to Purchaser shall be paid free and clear of any deduction or withholding on account of Taxes, and Purchaser shall not be entitled to deduct from the Purchase Price or any such other payment hereunder (or any portion thereof) any Transfer Taxes or other Taxes. Notwithstanding the foregoing, if any withholding or deduction is required on account of Taxes under applicable Law, then Purchaser or its paying agent shall pay an additional amount to Seller such that after accounting for any such withholding or deduction (including any withholding or deduction required on additional amounts paid) Seller receives the same amount that Seller would have received in the absence of any withholding or deduction. Purchaser or its paying agent shall promptly remit any amount it is required to withhold to the applicable Taxing Authority and furnish to Seller proof of remittance of the amount withheld and deducted. Seller shall, and shall cause each Selling Affiliate, to use commercially reasonable efforts to cooperate with Purchaser to obtain any legally available Tax exemption or to furnish to Purchaser any Tax certificate or documentation that Seller is legally permitted to furnish to Purchaser and that will permit Purchaser or its paying agent to reduce or eliminate any applicable withholding or deduction with respect to the payment of the Purchase Price or any other payment hereunder.
19
Section 2.11Indirect Taxes. The Purchase Price and any other payment required to be made pursuant to this Agreement are exclusive of any applicable Indirect Taxes. To the extent any Indirect Taxes are required to be paid to any Taxing Authority, Purchaser shall pay the amount of such Indirect Taxes to the Seller; provided, however, that Seller shall cooperate with Purchaser in claiming any available exemption or reduction of any Indirect Taxes. The Seller shall issue to Purchaser correct invoices in respect of such Indirect Taxes in accordance with applicable Law. If the actual amount (as finally determined) of an Indirect Tax properly chargeable on the relevant supply or service differs from the amount of Indirect Tax reflected on such invoices, the Parties shall fully cooperate and make appropriate adjustments to payments and invoices as required by applicable Law.
ARTICLE III
Representations and Warranties of the Seller
Except as set forth in the Seller Schedule, which Seller Schedule shall be organized into sections corresponding to the Sections (or, if applicable, subsections) of this Article III (provided, that any disclosure in a Section or subsection of the Seller Schedule shall apply to the corresponding Section or subsection of this Article III, as well as to the matters represented or warranted in such other Sections or subsections of this Article III with respect to which it is reasonably apparent on the face of such disclosure that such disclosure would apply or qualify), the Seller hereby represents and warrants to Purchaser as of the Execution Date and as of the Closing Date as follows:
Section 3.1Organization and Standing. The Seller is duly organized, validly existing and in good standing under the laws of the jurisdiction of its organization. The Seller has all requisite corporate or similar power and authority to own, lease, operate or otherwise hold the Purchased Assets owned, leased, operated or otherwise held by it and to carry on the Business as presently conducted. The Seller is duly qualified or authorized to do business as a foreign corporation in, and is in good standing under the Laws of, each jurisdiction in which it owns or leases real property and each other jurisdiction in which the conduct of its business or the ownership of its properties requires such qualification or authorization, except where the failure to be so qualified, authorized or in good standing would not have a Business Material Adverse Effect.
(b)Section 1.1(k) of the Seller Schedule contains a complete, true and correct list of the Selling Affiliates. Each Selling Affiliate that has title to any Purchased Assets is duly organized, validly existing, and in good standing under the laws of its jurisdiction of organization. Such Selling Affiliate has all requisite corporate or similar power and authority to own, lease, operate or otherwise hold its properties and assets and to carry on its portion of the Business as presently conducted and is duly qualified to do business and is in good standing as a foreign corporation or other entity in each jurisdiction where the ownership or operation of the Purchased Assets or the conduct of the Business requires such qualification, except where the failure to be so qualified, authorized or in good standing would not have a Business Material Adverse Effect.
Section 3.2Authority; Execution and Delivery; Enforceability. The Seller has all requisite corporate power and authority to execute this Agreement and the Ancillary Agreements
20
to which it is or will be a party and to consummate the transactions contemplated to be consummated by it under this Agreement and such Ancillary Agreements. The Seller has taken all corporate action required by its organizational documents to authorize the execution and delivery of this Agreement and the Ancillary Agreements to which it is or will be a party and to authorize the consummation of the transactions contemplated to be consummated by it under this Agreement and such Ancillary Agreements. The Seller has duly executed and delivered this Agreement and at the Closing will have duly executed and delivered each Ancillary Agreement to which it will be a party, and (assuming the due authorization, execution and delivery by each other Party) this Agreement constitutes, and (assuming the due authorization, execution and delivery by each other party thereto) each Ancillary Agreement to which it will be a party will after the Closing constitute, its legal, valid and binding obligation, enforceable against it in accordance with its terms subject, as to enforcement, to applicable bankruptcy, insolvency, moratorium, reorganization, fraudulent conveyance or similar Laws of general application affecting or relating to the enforcement of creditors’ rights generally and subject to equitable principles of general applicability (whether considered in an Action in equity or at law) (the “Enforceability Exceptions”).
(b)Each Selling Affiliate that will enter into an Ancillary Agreement has the requisite corporate or other entity power and authority to enter into and, perform its obligations under, each Ancillary Agreement to which it will be a party and to consummate the transactions contemplated thereby. The execution and delivery of the Ancillary Agreements to which any Selling Affiliate will be a party and the consummation of the transactions contemplated thereby have been duly authorized by all necessary corporate or other entity actions of such Selling Affiliate. Each Ancillary Agreement, when executed and delivered by a Selling Affiliate that is a party thereto, will constitute, in each case, assuming the due execution and delivery by each other party thereto, the valid and legally binding obligation of such Selling Affiliate, enforceable against such Selling Affiliate in accordance with its terms, subject to the Enforceability Exceptions.
Section 3.3Non-Contravention and Approvals. The execution and delivery by the Seller of this Agreement does not, the execution and delivery by the Seller and each Selling Affiliate of each Ancillary Agreement to which it will be a party will not, and the consummation by the Seller and each Selling Affiliate of the Acquisition and the other transactions contemplated to be consummated by it by this Agreement and such Ancillary Agreements will not, (i) conflict with or violate the certificate of incorporation or bylaws, or comparable organizational documents (“Governance Documents”), of Seller or any Selling Affiliate, (ii) except as set forth on Section 3.3(a)(ii) of the Seller Schedule, materially violate, conflict with, breach, result in a breach of, or constitute a default under or result in the termination of any Material Contract or any Contract to which Seller or any of its Affiliates is a party or by which it is bound and which has been filed (including by incorporation by reference) with the SEC as an exhibit to Seller’s most recent Form 10-K, Form 10-Q or any 8-K filed since the most recent 10-Q, in each case, filed with the SEC, (iii) conflict with or violate any judgment, order or decree (“Judgment”) or Law applicable to the Seller, any Selling Affiliate, the Business or the Purchased Assets, or (iv) result in the creation of any material Lien (other than Permitted Liens or Liens arising from acts of Purchaser or its Affiliates) on any of the Purchased Assets.
21
(b)Except for (i) compliance with and filings under the HSR Act, (ii) compliance with and filings, notifications and approvals under any other antitrust, competition or trade regulation Laws set forth on Section 3.3(b)(ii) of the Seller Schedule (iii) consents, permits, authorizations, declarations, filings or registrations that have become applicable solely as a result of the specific regulatory status of Purchaser or its Affiliates and (iv) items set forth in Section 3.3(b)(iv) of the Seller Schedule, no notice to, filing with, permit of, authorization of, exemption by, or consent of, any Governmental Authority is required for the Seller and the Selling Affiliates to consummate the Acquisition and the other transactions contemplated hereby or by the Ancillary Agreements.
Section 3.4Title; Sufficiency of Purchased Assets; Personal Property.
(a)The Seller or a Selling Affiliate owns and has good and valid title to all Purchased Assets, free and clear of all Liens (other than Permitted Liens).
(b)Except as set forth on Section 3.4(b) of the Seller Schedule, (i) the Purchased Assets (assuming the receipt of all Approvals), (ii) the Seller’s rights under the Shared Contracts, (iii) the services being provided to Purchaser contemplated by the Transitional Services Agreement and the other Ancillary Agreements, (iv) the Intellectual Property licensed to Purchaser under the Transitional Trademark License Agreement, (v) the Business Employees, and (vi) real property and general corporate, finance and support services (including quality, pharmacovigilance and supply chain management services) and functions provided by Seller and its Affiliates to the Business prior to the Closing, including employees providing such services, constitute all of the material properties, rights, interests and other tangible and intangible assets used or held for use by Seller and its Affiliates in connection with the Business and would be sufficient for Purchaser to conduct the Business from and after the Closing as conducted immediately prior to the Closing, in all material respects, by the Seller and the Selling Affiliates.
(c)Section 3.4(c) of the Seller Schedule sets forth a true, correct and complete list of all leases of cars used primarily in the Business (the “Personal Property Leases”). All of the items of personal property under the Personal Property Leases and included in the Purchased Assets are in good condition and repair (ordinary wear and tear excepted) and are suitable for the purposes used, and, as applicable, such property is in all material respects in the condition required of such property by the terms of the lease applicable thereto during the term of the lease.
Section 3.5Real Property. There is no material real property or interests in real property owned, leased by, licensed to, occupied or subleased by the Seller or any Selling Affiliate that is used or held for use exclusively in the operation or conduct of the Business.
Section 3.6Intellectual Property. The Seller or a Selling Affiliate is the sole and exclusive owner of the Purchased Intellectual Property and the Purchased Domain Names. The Purchased Intellectual Property was developed without funding or other contributions from a government, nonprofit or academic organization, and no government, nonprofit or academic organization has any rights in or to the Purchased Intellectual Property. The Patents and Trademarks included in the Purchased Intellectual Property, together with the Purchased Domain Names and the Intellectual Property licensed to Purchaser under the Transitional Trademark License Agreement, constitute all of the material Patents, Know-how, Trademarks and Domain
22
Names reasonably necessary for the conduct of the Business as conducted by the Seller immediately prior to the Execution Date. Section 3.6(a) of the Seller Schedule sets forth, as of the date hereof, a true and complete list of all Purchased Intellectual Property owned by Seller or one of its Affiliates that has not expired or been abandoned and has issued, been registered or granted or that is the subject of an application for registration, issuance or grant (“Owned Registered Product IP”). Sellers have received no written notice challenging the validity, enforceability or good standing of any Purchased Intellectual Property and all required maintenance fees, annuity fees or renewal fees for the Owned Registered Product IP that are due and payable prior to the Closing Date have been or will be paid prior to the Closing Date.
(b)Except with respect to non-exclusive licenses and authorizations to use granted to or by third parties in the ordinary course of business or as otherwise contemplated by this Agreement, Section 3.6(b) of the Seller Schedule lists, as of the Execution Date, all of the Contracts (i) pursuant to which the Seller and the Selling Affiliates obtained the right to use or practice rights under third party Registered Intellectual Property that are exclusively related to the Product or the Business and that are material to the operation of the Business or (ii) pursuant to which the Seller or any Selling Affiliate has granted a third party a license to use or practice under any Patent or Trademark that is material to the operation of the Business and included in the Purchased Intellectual Property.
(c)Except as would not, individually or in the aggregate, reasonably be expected to be material to the Business, as of the Execution Date, to the Knowledge of the Seller there are no (i) adverse third party actions or claims pending or existing against, or otherwise known to the Seller or any of the Selling Affiliates by any Person in any court or arbitration or by or before any Governmental Authority, or (ii) adverse third party allegations made to the Seller, in any such case (clauses (i) and (ii)) to the effect that the operation or conduct of the Business infringes or misappropriates the Intellectual Property of such Person, or that challenges the validity or enforceability of any Purchased Intellectual Property. Purchaser acknowledges and agrees that the representations and warranties set forth in this Section 3.6(c) are the only representations and warranties the Seller make in this Agreement with respect to any activity by the Seller or the Selling Affiliates that constitutes or may constitute infringement, misappropriation or other violation of any Intellectual Property.
(d)Except as set forth in Section 3.6(d) of the Seller Schedule, none of the Patents or Trademarks in the Purchased Intellectual Property is involved in any interference, reissue, reexamination, derivation, supplemental examination, inter partes review, post-grant review, conflict, opposition, cancellation, litigation or other post-issuance proceeding, and, to the Knowledge of the Seller, there has been no threat or other written indication that any such proceeding shall hereafter be commenced.
(e)Except as set forth in Section 3.6(e) of the Seller Schedule, as of the Execution Date, (i) there are no claims pending or threatened by, or otherwise known to, the Seller or any of the Selling Affiliates against, or that to the Knowledge of the Seller reasonably could be asserted against, any Person in writing, nor has the Seller or any Selling Affiliate sent any written notice to any Person, regarding any actual or potential infringement, dilution, misappropriation or other unauthorized use of any of the Purchased Intellectual Property and (ii) to the Knowledge of the Seller, there are no such infringements, dilutions, misappropriations
23
or other unauthorized uses of such Purchased Intellectual Property by any Person, other than in the case of clauses (i) and (ii), as would not, individually or in the aggregate, reasonably be expected to be material to the Business.
(f)The Purchased Intellectual Property is free of Liens and is not subject to any orders, judgments, or limitation or restrictions on use or otherwise. The Seller is not a party to any action, suit, or proceeding that prohibits or restricts the exploitation of the Purchased Intellectual Property, or that restricts in any manner the use, transfer or licensing thereof by the Seller or may affect the validity or enforceability of the Purchased Intellectual Property.
(g)All Purchased Intellectual Property and the rights to any inventions claimed or disclosed therein, have been or will be properly assigned to the Seller, and all such assignments have been or will be properly recorded in the United States Patent and Trademark Office (with respect to all patent rights in the Purchased Intellectual Property in the United States) or to any analogous foreign Governmental Authority prior to the Closing Date.1
(h)Seller has and will have the full right, power and authority to assign and will assign its right, title and interest in the Purchased Intellectual Property to the Purchaser as set forth in this Agreement, the Ancillary Agreements and the transactions contemplated hereby and thereby. Immediately after the Closing, Purchaser shall have the same ownership rights in the Purchased Intellectual Property that the Seller had immediately prior to the Closing. Neither the execution, delivery or performance of the Agreement nor the consummation of the transactions contemplated hereby will: (i) contravene, conflict with or result in any limitation on the Seller’s right, title or interest in or to any of the Purchased Intellectual Property (except as contemplated by this Agreement); (ii) result in the release, disclosure, forfeiture or delivery of any Purchased Intellectual Property by or to any other Person; or (iii) cause the grant, assignment or transfer to any other Person of any license or other right or interest under, to or in any of the Purchased Intellectual Property (except as contemplated by this Agreement).
(i)The Seller has taken commercially reasonable measures to protect and maintain the proprietary nature of each item of Purchased Intellectual Property. Except as set forth in Section 3.6(i) of the Seller Schedule, all current and former employees, consultants and contractors of the Seller involved in the creation or development of any Purchased Intellectual Property have irrevocably assigned all of their respective right, title and interest in and to such Purchased Intellectual Property to the Seller and are bound by confidentiality obligations through signed agreements containing valid intellectual property assignments and confidentiality provisions in favor of the Seller consistent with best practices in the industry in which the Seller operates and in a form provided to the Purchaser. The Seller does not owe any ongoing or contingent compensation or remuneration (other than wages, salary and benefits payable to employees, and hourly fees, time and materials fees, support fees, milestone payments for services performed or to be performed, set fees or other non-continuing fees for services payable to contractors or consultants, for work performed) to a current or former service provider or other consultant or contractor (including any research facility or university) involved in the use or other exploitation of any Purchased Intellectual Property.
1 | Note to Seller: Please disclose any IP that is not held by Seller as of the Execution Date. |
24
(j)No employee or former employee or any present or former Affiliate of the Seller has any right, title, or interest, directly or indirectly, in whole or in part, in any Purchased Intellectual Property. No employer or former employer of any employee (including any research facility or university) has any claim, right (whether or not currently exercisable) or interest to or in any material Purchased Intellectual Property or has excluded any Intellectual Property from any assignment agreement referenced above that is used in or necessary for the Subject Products.
Section 3.7Contracts.
(a)Section 3.7(a) of the Seller Schedule sets forth all of the Material Contracts which constitute Purchased Contracts.
(b)Each of the Purchased Contracts, excluding those which by their terms, expire prior to the Closing Date, is in effect and constitutes a legal, valid and binding agreement of one or more of the Seller or any Selling Affiliates, and to the Knowledge of the Seller, each other party thereto, enforceable in accordance with its terms, subject to the Enforceability Exceptions. Neither the Seller or applicable Selling Affiliate party to any Purchased Contract nor, to the Knowledge of the Seller, any other party to such Contract, is in breach or default under such Contract in any material respect, and no event has occurred that with notice or lapse of time or both, would constitute a default or breach under any Purchased Contract. None of the Seller or any Selling Affiliate has given any written notice to any third party that is a party to any Purchased Contract that it intends to terminate such Purchased Contract and none of the Seller or any Selling Affiliate has received any written notice from a third party stating that such third party intends to terminate any material Purchased Contract, in each case, other than valid termination at the end of the term of such Purchased Contract or otherwise in the ordinary course of business. True and complete copies of all Purchased Contracts have been made available to Purchaser, except to the extent such Purchased Contracts have been redacted to (a) enable compliance with Laws relating to antitrust or the safeguarding of data privacy, (b) comply with confidentiality obligations owed to third parties or (c) exclude information not related to the Business or the Product.
(c)None of the agreements set forth in Annex 2.1(a) of the Seller Schedule substantially deviates from the forms of Discount Pricing Agreements made available to Purchaser.
Section 3.8Taxes. To the extent relevant for determining any liability for Taxes for which Purchaser or its Affiliates would be responsible:
(a)There are no material Liens for Taxes on any of the Purchased Assets or the Business (other than Permitted Liens); and
(b)Seller has materially complied with all applicable Laws relating to the collection, reporting and remittance of gross withholding Taxes with respect to the Purchased Assets or the Business.
Section 3.9Litigation. (i) There currently is no Action pending or, to the Knowledge of the Seller, threatened in writing against the Seller or any of the Selling Affiliates or their respective assets by or before any Governmental Authority, on behalf of any Governmental
25
Authority or by a third party seeking to assert a claim, in each case, relating primarily to the Business or the Purchased Assets, and (ii) there is no Judgment of a Governmental Authority to which the Seller or any of the Selling Affiliates is subject relating primarily to the Business or the Purchased Assets.
(b)There exists no Action which prohibits, restricts or seeks to enjoin the transactions contemplated by this Agreement or the Ancillary Agreements.
Section 3.10Employee Matters.
(a)Section 3.10(a) of the Seller Schedule contains a complete and accurate list of each material Business Benefit Plan. Prior to the Execution Date, complete and accurate copies of the plan document, if any, for each Business Benefit Plan, including any amendments to the plan document have been made available to Purchaser or, if such plan is unwritten, a written summary of such plan’s material terms.
(b)Each Business Benefit Plan has been maintained, operated and administered in compliance with applicable Laws and with the terms of such Business Benefit Plan, except where the failure to comply would not reasonably be expected to result in material Liability to Purchaser. No Business Benefit Plan is, and none of the Seller or any of its ERISA affiliates has, during the preceding five-year period, contributed to, or been obligated to contribute to, or otherwise had any obligation or liability in connection with any benefit plan that is subject to Section 302 or Title IV of ERISA, or Section 412 or 430 of the Code or is otherwise a defined benefit pension plan, including a “multiemployer plan” (within the meaning of Sections 3(37) or 4001(a)(3) of ERISA). There are no pending, or to the Knowledge of the Seller, threatened, investigations by any Governmental Authority with respect to or termination proceedings or other claims, suits or proceedings (except routine claims for benefits payable in the ordinary course) against or involving any Business Benefit Plan, except as would not reasonably be expected to result in any material Liability to Purchaser.
(c)Neither the execution and delivery of this Agreement nor the consummation of the transactions contemplated hereby will (i) result in any payment becoming due to any Business Employee, (ii) increase any benefits otherwise payable to any Business Employee under any Business Benefit Plan, (iii) result in the acceleration of the time of payment, funding, or vesting of any benefits under any Business Benefit Plan or (iv) give rise to any “parachute payment” (within the meaning of Section 280G(b)(2) of the Code) to be made to any Business Employee.
(d)The Seller and each Selling Affiliate is, and for the past three years has been, in compliance with all Laws governing employment and labor, including all contractual commitments and all such Laws relating to wages, hours, collective bargaining, discrimination, harassment, retaliation, immigration, civil rights, safety and health, and workers’ classification and compensation, to the extent such Laws apply to the Seller or any Selling Affiliate with respect to the Business Employees, except for instances of noncompliance that would not reasonably be expected to result in material Liability to Purchaser. To the Knowledge of the Seller, neither Seller nor any Selling Affiliate is liable for any arrears of wages, other
26
compensation or benefits, or any Taxes or penalties for failure to comply with any of the foregoing, in each case with respect to the Business Employees.
(e)There are (i) no unions or other collective bargaining units or employee organizing entities certified as representing any of the Business Employees or holding bargaining rights with respect to the Business Employees, (ii) no unions or other collective bargaining units or employee organizing entities recognized by the Seller or any Selling Affiliate as representing any of the Business Employees and (iii) no pending, or to the Knowledge of the Seller, threatened union organizing activities, controversies, strikes, slowdowns, or any other material labor disputes, involving any Business Employees. There are no pending, or to the Knowledge of the Seller, threatened, grievances or unfair labor practice complaints against Seller or any Selling Affiliates before the National Labor Relations Board or any other Governmental Authority or otherwise with respect to any Business Employee.
(f)True and correct information has been made available to Purchaser regarding the following with respect to each Business Employee: (i) name, job title and employing entity, (ii) original hire date and service date (if different), (iii) principal location of employment, (iv) employment status (including exempt or non-exempt, full-time or part-time, or temporary), (v) leave status and anticipated date of return to full-service; (vi) current base salary or wage rate, (vii) target bonus or commission paid for the fiscal year ending December 31, 2021 and bonus or commission opportunity for the fiscal year commencing January 1, 2022, (viii) any material benefits, whether variable or fixed, (ix) vacation and sick leave entitlement and accrual, (x) confirmation of eligibility to work in the applicable jurisdiction and visa status and (xi) information relating to any, past, current or pending disciplinary actions with respect to any such Business Employee. As of the date of this Agreement, no Business Employee has given notice of terminating his or her employment or is under notice of dismissal. No Business Employee who receives an offer of employment with the Required Terms pursuant to Section 6.11(a) requires or will require a visa, work permit or employment pass or other similar approval in connection with his or her employment with Purchaser (or any Affiliate of Purchaser).
(g)Except as set forth in Section 3.10(g) of the Seller Schedule, other than the Business Employees, neither the Seller nor any Selling Affiliate employs any individual who performs sales, marketing, quality assurance, manufacturing or regulatory functions primarily or exclusively for the Business. Each Business Employee has been primarily dedicated to the Business since December 31, 2020 (or, for any such Business Employee hired after December 31, 2020, since the date of such Business Employee’s hire). Except as set forth in Section 3.10(g) of the Seller Schedule, since December 31, 2020, no employee of Seller or any Selling Affiliate who was primarily dedicated to the Business has been transferred to any division or business unit of Seller other than the Business.
Section 3.11Compliance with Applicable Laws.
(a)The Seller and the Selling Affiliates, with respect to the operation of the Business, are and during the three-year period immediately preceding the Execution Date have been, in compliance in all material respects with all Laws and Permits applicable to the Business and the Purchased Assets, including Environmental Laws, Laws relating to the safeguarding of data privacy, Anti-Corruption Laws, economic sanctions Laws, export control Laws and all
27
national, provincial, state, local, municipal or foreign Laws concerning the important of merchandise, the terms and conduct of international transactions and making or receiving international payments.
(b)During the three-year period immediately preceding the Execution Date, the Seller and the Selling Affiliates, with respect to operation of the Business, have not received a subpoena, letter of investigation, or other document from a Governmental Authority or a private party seeking to assert a claim on its behalf alleging a violation of any such Laws.
(c)This Section 3.11 does not relate to (i) Tax, which is the subject of Section 3.8, (ii) employment and employee benefits matters, which are the subject of Section 3.10 and (iii) Regulatory Laws, which are the subject of Section 3.12. Entry by Purchaser and Parent Guarantor into this Agreement or the Ancillary Agreements will not cause Purchaser or its Affiliates (which shall, solely for this purpose, include the CVC Network) to either (i) transact with any person on a sanctions list maintained by OFAC, or (ii) breach any prohibition imposed by the Annex to the United States Executive Order 13224, the USA PATRIOT Act, the Trading with the Enemy Act or OFAC regulations.
Section 3.12Regulatory Compliance.
(a)The Seller and the Selling Affiliates are conducting, and during the three-year period immediately preceding the Execution Date have conducted, the Business in compliance in all material respects with all applicable Regulatory Laws governing the Exploitation of the Product. During the three-year period immediately preceding the Execution Date, none of the Seller or any of the Selling Affiliates has received any notice in writing or otherwise, including any Warning Letter, Untitled Letter, FDA Form-483, adverse inspection finding, penalty, fine, sanction, assessment, written request for corrective or remedial action, or notice of violation letter by any Governmental Authority (“Notice”), which has, or reasonably should have, led the Seller to determine that Seller, any of the Selling Affiliates, the Product, or the Business was not in compliance with any Regulatory Law.
(b)Except as set forth in Section 3.12(b)(i) of the Seller Schedule, the Seller and the Selling Affiliates possess all Regulatory Approvals and Permits necessary to conduct the Business as currently conducted, and a true and correct list of such Regulatory Approvals and Permits is set forth in Section 3.12(b)(ii) of the Seller Schedule. Such Regulatory Approvals and Permits are valid and in full force and effect. None of the Seller or any Selling Affiliate is, or since three years prior to the Execution Date has been, in material non-compliance with the terms of any such Regulatory Approval or Permit or has received any communication or notice of enforcement from any Governmental Authority relating to any elements or facts that may undermine the ongoing validity of any such Regulatory Approval or Permit. There are no Actions or inspections pending or, to the Knowledge of the Seller, threatened that could result in the suspension, cancellation, revocation, termination, or material modification of any such Regulatory Approvals or Permits. With respect to the Business and the Product, the Seller and each Selling Affiliate have filed with the relevant Governmental Authorities all Regulatory Approvals and Permits and material requests, notices, information, and annual or other reports, including adverse event reports, each of which, including any material updates, changes,
28
corrections or modifications thereto, were, to the Knowledge of the Seller, true, complete, and correct in all material respects as of the date of submission.
(c)None of the Seller, any Selling Affiliate or, to the Knowledge of the Seller, any third party engaged by the Seller in connection with the Exploitation of the Product in any jurisdiction has received any notice of enforcement action from any applicable Governmental Authority in the three years immediately prior to the Execution Date with respect to any facility engaged in the Exploitation of such Product or has Exploited the Product in material non-compliance with any applicable Regulatory Law. During the three-year period immediately preceding the Execution Date, there have been no voluntary or required recalls or market withdrawals, seizures, detentions, import alerts or similar actions, or any material quality, safety, efficacy or performance issues, defects, or complaints with respect to the Product and no such matters are pending, outstanding, or, to the Knowledge of the Seller, threatened.
(d)All Manufacturing operations conducted by or on behalf of the Seller and its Affiliates involving the Product, including contract manufacturers engaged by the Seller to Manufacture the Product, have been and are being conducted in accordance, in all material respects, with all applicable Regulatory Laws, including GMP. All such manufacturing operations are in material compliance with all applicable registration and listing requirements set forth in 21 U.S.C. Section 360 and 21 C.F.R. Part 207 and all similar Laws in those jurisdictions in which human clinical trials or manufacturing activities have been or are being conducted by the Seller and its Affiliates involving the Product. All non-clinical, pre-clinical and clinical trials, clinical investigations and performance evaluations conducted by or on behalf of the Seller and its Affiliates involving the Product are being and have been conducted in material compliance with all applicable protocols and Regulatory Laws and no such trials, investigations, or evaluations have been subject to any clinical hold, or any other Action requiring or requesting the termination, suspension, or material modification of such trials, investigations, or evaluations by any Governmental Authority and no such matters are pending.
(e)None of the Seller, any of its Affiliates or any of their respective directors or employees and, to the Knowledge of the Seller, no contractors, agents, or representatives acting for the Seller or its Affiliates, has committed any act, made any statement or failed to make any statement, relating to the Product or the development or manufacturing thereof that would reasonably be expected to provide a basis for FDA to invoke its policy with respect to “Fraud, Untrue Statements of Material Facts, Bribery, and Illegal Gratuities” set forth in 56 Fed. Reg. 46191 (September 10, 1991) and any amendments thereto or to provide a basis for a foreign Governmental Authority to invoke a comparable foreign policy.
(f)None of the Seller, any of its Affiliates or any of their respective officers, directors, or employees or, to the Knowledge of the Seller, any contractor, agent, or representative acting for the Seller or its Affiliates in connection with the Business is or ever has been debarred, excluded, or suspended under any applicable Regulatory Law, or from participation, or otherwise been deemed ineligible to participate, in any health care programs of any Governmental Authority, or convicted of any crime regarding health care products or services, or engaged in any conduct that would reasonably be expected to result in any such debarment, exclusion, suspension, or ineligibility, including (i) debarment under 21 U.S.C.
29
Section 335a or any similar state or foreign Law and (ii) exclusion under 42 U.S.C. Section 1320a–7 or any similar state or foreign Law.
(g)There are no outstanding material compliance complaints or reports, internal compliance investigations, or compliance corrective actions relating to the Business.
Section 3.13Brokers and Finders. There is no investment banker, broker, finder, financial advisor or other intermediary entitled to any fee or commission payable by Seller, any Selling Affiliate, Purchaser or any of its Affiliates in connection with the transactions contemplated by this Agreement or the Ancillary Agreements.
Section 3.14Absence of Certain Changes. During the three-year period immediately preceding the Execution Date, except as disclosed in Seller’s public filings, (a) the Business has been conducted in the ordinary course of business consistent with past practice and (b) there has not been any event, occurrence, development or state of circumstances or facts that, individually or in the aggregate, has had a Business Material Adverse Effect.
Section 3.15Solvency. Upon consummation of the Acquisition, and after giving effect to the transactions contemplated hereby, including payment of all amounts required to be paid in connection with the consummation of the transactions contemplated hereby, and payment of all related fees and expenses, each of Seller and its Affiliates will be Solvent as of the Closing and immediately after the consummation of the transactions contemplated hereby. The transfer of the Purchased Assets is not being made and no obligation is being incurred by Seller in connection with the Acquisition with the intent to hinder, delay or defraud either present or future creditors of Seller or any of its Affiliates.
ARTICLE IV
Representations and Warranties of Purchaser
Except as set forth in the Purchaser Schedule, which Purchaser Schedule shall be organized into sections corresponding to the Sections (or, if applicable, subsections) of this Article IV (provided, that any disclosure in a Section or subsection of the Purchaser Schedule shall apply to the corresponding Section or subsection of this Article IV, as well as to the matters represented or warranted in such other Sections or subsections of this Article IV with respect to which it is reasonably apparent on the face of such disclosure that such disclosure would apply or qualify), Purchaser hereby represents and warrants to the Seller as of the Execution Date and as of the Closing Date as follows:
Section 4.1Organization. Purchaser, and each Affiliate of Purchaser that will enter into any Ancillary Agreement, is a legal entity duly organized, validly existing and in good standing under the Laws of the jurisdiction of its organization or incorporation.
Section 4.2Authority; Execution and Delivery; Enforceability.
(a)Purchaser has the requisite corporate or other entity power and authority to enter into this Agreement and the Ancillary Agreements to which it is a party, to perform its obligations hereunder and thereunder and to consummate the transactions contemplated hereby and thereby. The execution and delivery of this Agreement and Ancillary Agreements to which
30
Purchaser is a party and the consummation of the transactions contemplated hereby and thereby have been duly authorized by the necessary corporate or other entity actions of Purchaser. Purchaser has duly executed and delivered this Agreement and at the Closing will have duly executed and delivered each Ancillary Agreement to which it will be a party. This Agreement constitutes and each Ancillary Agreement to which Purchaser will be a party, when executed and delivered by Purchaser, will constitute, in each case, assuming the due execution and delivery by each other party thereto, the valid and legally binding obligation of Purchaser, enforceable against Purchaser in accordance with its terms, subject to the Enforceability Exceptions.
(b)Each Affiliate of Purchaser that will enter into an Ancillary Agreement has the corporate or other entity power and authority to enter into and, perform its obligations under, each Ancillary Agreement to which it will be a party and to consummate the transactions contemplated thereby. The execution and delivery of the Ancillary Agreements to which any Affiliate of Purchaser will be a party and the consummation of the transactions contemplated thereby have been duly authorized by all necessary corporate or other entity actions of such Affiliate. Each Ancillary Agreement, when executed and delivered by an Affiliate of Purchaser that is a party thereto, will constitute, in each case, assuming the due execution and delivery by each other party thereto, the valid and legally binding obligation of such Affiliate, enforceable against such Affiliate in accordance with its terms, subject to the Enforceability Exceptions.
Section 4.3Non-Contravention and Approvals.
(a)The execution, delivery and performance by Purchaser of this Agreement and of each Ancillary Agreement to which it will be a party and the execution, delivery and performance by each Affiliate of Purchaser of each Ancillary Agreement to which such Affiliate will be a party do not and will not (i) violate the certificate of incorporation or bylaws, or comparable organization documents, of Purchaser or such Affiliate, as applicable, (ii) violate any Law applicable to Purchaser or such Affiliate, as applicable or (iii) violate, breach or constitute a default under or result in the termination of any material Contract to which Purchaser or such Affiliate is a party, except, in the case of (ii) or (iii), for such violations, breaches, defaults or terminations that would not reasonably be expected to have a Purchaser Material Adverse Effect.
(b)Except for (i) compliance with and filings under the HSR Act, (ii) consents, permits or authorizations that if not received, or declarations, filings or registrations that if not made, would not reasonably be expected to have a Purchaser Material Adverse Effect, and (iii) compliance with and filings, notifications and approvals under any antitrust, competition, trade regulation or foreign investment Laws set forth on Section 4.3(b) of the Purchaser Schedule (the “Foreign Merger Control Laws”), no notice to, filing with, permit of, authorization of, exemption by, or consent of, Governmental Authority or other Person is required for Purchaser to consummate the transactions contemplated hereby or by the Ancillary Agreements.
Section 4.4No Litigation. There is no Action pending or, to the Knowledge of Purchaser, threatened against Purchaser or any of its Affiliates before any Governmental Authority and there is no Judgment of a Governmental Authority to which Purchaser or any of its Affiliates is subject, except, for any Action, Judgment that would not reasonably be expected to have a Purchaser Material Adverse Effect.
31
Section 4.5Exclusivity of Representations; Independent Investigation.
(a)Purchaser acknowledges and agrees that, other than the representations and warranties specifically contained in this Agreement or in the Ancillary Agreements there are no representations or warranties of the Seller, any of the Selling Affiliates or any other Person either expressed, statutory or implied with respect to the Seller, its Affiliates, the Purchased Assets, the Assumed Liabilities or the Business, including with respect to any of their respective rights or assets, or the transactions contemplated hereby, individually or collectively. Without limiting the generality of the foregoing, (i) Purchaser acknowledges and agrees that none of the Seller, the Selling Affiliates or their other Affiliates or respective representatives makes any representations or warranties relating to (1) the maintenance, repair, condition, design, performance or marketability of any Purchased Asset including with respect to fitness for a particular purpose, (2) the operation of the Purchased Assets or the Business after the Closing (3) the probable success or profitability of the Purchased Assets or the Business, or (4) the availability, amount or likelihood of any Separate Payment and (ii) Purchaser understands the competitive dynamics and risks inherent in the pharmaceutical industry, including the potential for development, authorization, approval or market entry of products that may compete with any Subject Product.
(b)Purchaser acknowledges that it, its Affiliates and their respective representatives have been permitted full access to the books and records, facilities, equipment, personnel, Contracts and other properties and assets of the Business that it, its Affiliates and their respective representatives have desired or requested to see and review, and that it and its Affiliates and their respective representatives have had a full opportunity to meet with the officers and employees of the Seller to discuss the Business. Except as expressly set forth in any representation or warranty of the Seller set forth in Article III or in any Ancillary Agreement, Purchaser acknowledges and agrees that no Person, including the Purchaser Indemnitees, shall have any claim (whether in warranty, contract, tort (including negligence, strict liability or innocent or negligent misrepresentation or misstatement) or otherwise) or right to indemnification pursuant to Article IX (or otherwise) with respect to any information, summaries, documents or materials made available or otherwise furnished to or for Purchaser, its Affiliates or their respective representatives by the Seller, any of the Selling Affiliates, or any of their other Affiliates or respective representatives.
(c)Purchaser, its Affiliates and their respective representatives have received and may continue to receive from the Seller, the Selling Affiliates and their other Affiliates and respective representatives certain estimates, projections and other forecasts for the Business and certain plan and budget information. Purchaser acknowledges that these estimates, projections, forecasts, plans and budgets, and the assumptions on which they are based, were prepared for specific purposes and may vary significantly from each other. Further, Purchaser acknowledges that there are uncertainties inherent in attempting to make such estimates, projections, forecasts, plans and budgets, that Purchaser is taking full responsibility for making its own evaluation of the adequacy and accuracy of all estimates, projections, forecasts, plans and budgets so furnished to it, its Affiliates or their respective representatives (including the reasonableness of the assumptions underlying such estimates, projections, forecasts, plans and budgets) and that Purchaser is not relying on any estimates, projections, forecasts, plans or budgets made available or otherwise furnished by the Seller, the Selling Affiliates or their other Affiliates or respective
32
representatives, and Purchaser shall not, and shall cause its controlled Affiliates and their respective representatives not to, hold any such Person liable with respect thereto (whether in warranty, contract, tort (including negligence, strict liability or innocent or negligent misrepresentation or misstatement) or otherwise).
Section 4.6Solvency. From and following consummation of the Acquisition, and after giving effect to the transactions contemplated hereby, including the payment of the Purchase Price, payment of all amounts required to be paid in connection with the consummation of the transactions contemplated hereby (including all Additional Payments), and payment of all related fees and expenses, Purchaser will be Solvent.
Section 4.7Brokers and Finders. There is no investment banker, broker, finder, financial advisor or other intermediary that might be entitled to any fee or commission payable by the Seller or any of its Affiliates in connection with the transactions contemplated by this Agreement or the Ancillary Agreements.
Section 4.8Debarment. None of Purchaser, any of its Subsidiaries or any of their respective directors or employees or, to the Knowledge of Purchaser, any representative acting for Purchaser or its Subsidiaries is or ever has been debarred, excluded, or suspended from participation, or otherwise been deemed ineligible to participate, in any health care programs of any Governmental Authority, or convicted of any crime regarding health care products or services, or engaged in any conduct that would reasonably be expected to result in any such debarment, exclusion, suspension, or ineligibility, including (a) debarment under 21 U.S.C. Section 335a or any similar state or foreign Law and (b) exclusion under 42 U.S.C. Section 1320a–7 or any similar state or foreign Law. Financial Ability.
(a)Purchaser or its Affiliates, as applicable, has received and accepted a fully executed debt Commitment Letter, dated on or before the date hereof (together with all exhibits, annexes and schedules thereto, and as amended, supplemented or replaced in compliance with this Agreement, the “Debt Commitment Letter”) with the lenders named therein (collectively, the “Lenders”) pursuant to which such Lenders have agreed, subject to the terms and conditions thereof, to provide the amounts of debt financing set forth therein, and for the purposes described therein. The term debt financing committed pursuant to the Debt Commitment Letter is referred to in this Agreement as the “Debt Financing.”
(b)Purchaser has delivered to Seller a true, complete and correct copy of the executed Debt Commitment Letter save for redactions to delete any confidential compensation information, certain market flex provisions, fee amounts, pricing caps and securities demands (none of which would adversely affect the amount, conditionality, enforceability, termination or availability of the Debt Financing in any material respect). The Debt Commitment Letter constitutes the entire and complete agreement of the parties thereto with respect to the Debt Financing and as of the date of this Agreement there are no side letters or other letters, contracts or arrangements except for customary fee letters, engagement letters or other letters, contracts or arrangements which do not adversely affect the amount, conditionality, enforceability, termination, or availability of the Debt Financing in any material respect. Purchaser shall promptly deliver to the Seller copies of any amendment, supplement, modification or alternative Debt Commitment Letter.
33
(c)Except as expressly set forth in the Debt Commitment Letter, there are no conditions precedent to the obligations of the Lenders to provide the Debt Financing, or any terms or contingencies that would, or could reasonably be expected to, permit the Lenders to reduce the total amount of the Debt Financing. Assuming the satisfaction of the conditions in Section 7.1 and Section 7.2, as of the date of this Agreement, Purchaser does not have any reason to believe that it (or its relevant Affiliate(s)) will be unable to satisfy on a timely basis all terms and conditions to be satisfied by it (or its relevant Affiliate(s)) in the Debt Commitment Letter on or prior to the Closing Date, nor does Purchaser have any reason to believe that any of the Lenders will not perform their obligations thereunder.
(d)The Debt Financing, when funded in accordance with the Debt Commitment Letter, shall directly or indirectly provide Purchaser with cash proceeds sufficient, together with the resources of the Purchaser and its Affiliates, for the Purchaser to pay the Purchase Price. Purchaser has not incurred, and is not contemplating or aware of, any obligation, commitment, restriction or other Liability of any kind, in each case that would impair or adversely affect such resources, funds or capabilities.
(e)As of the date of this Agreement, the Debt Commitment Letter is (i) legal, valid, binding and enforceable obligations of Purchaser (or its relevant Affiliate(s)) and, to the Knowledge of Purchaser, of each of the other parties thereto in accordance with their respective terms (subject to the Enforceability Exceptions) and (ii) in full force and effect. As of the date of this Agreement, no event has occurred that, with or without notice, lapse of time, or both, would reasonably be expected to constitute a default or breach or a failure to satisfy a condition precedent on the part of Purchaser or, to the knowledge of Purchaser, the Lenders under the terms of the Debt Commitment Letter. Purchaser (or its relevant Affiliate) has irrevocably paid in full (or caused to be paid) any and all commitment fees or other fees and expenses required to be paid pursuant to the terms of the Debt Commitment Letter on or before the date of this Agreement and will continue to timely pay in full (or caused to be paid) any such amounts arising under the Debt Commitment Letter as and when they become due and payable. As of the date of this Agreement, the Debt Commitment Letter has not been modified, amended, withdrawn or restated and none of the commitments under the Debt Commitment Letter have been withdrawn, terminated or rescinded in any respect.
(f)In no event shall the receipt or availability of any funds or financing (including, for the avoidance of doubt, the Debt Financing) by Purchaser or any Affiliate or any other financing or other transactions be a condition to any of Purchaser’s obligations hereunder.
ARTICLE V
Representations and Warranties of Parent Guarantor
Parent Guarantor hereby represents and warrants to the Seller as of the Execution Date as follows:
Section 5.1Organization. Parent Guarantor is a legal entity duly organized, validly existing and in good standing under the Laws of the jurisdiction of its organization or incorporation.
34
Section 5.2Authority; Execution and Delivery; Enforceability. Parent Guarantor has the requisite corporate or other entity power and authority to enter into this Agreement and the Ancillary Agreements to which it is a party, to perform its obligations hereunder and thereunder and to consummate the transactions contemplated hereby and thereby. The execution and delivery of this Agreement and Ancillary Agreements to which Parent Guarantor is a party and the consummation of the transactions contemplated hereby and thereby have been duly authorized by the necessary corporate or other entity actions of Parent Guarantor. Parent Guarantor has duly executed and delivered this Agreement and at the Closing will have duly executed and delivered each Ancillary Agreement to which it will be a party. This Agreement constitutes and each Ancillary Agreement to which Parent Guarantor will be a party, when executed and delivered by Parent Guarantor, will constitute, in each case, assuming the due execution and delivery by each other party thereto, the valid and legally binding obligation of Parent Guarantor, enforceable against Parent Guarantor in accordance with its terms, subject to the Enforceability Exceptions.
ARTICLE VI
Covenants
Section 6.1Conduct of Business.
(a)Except for (i) matters set forth in Section 6.1(a) of the Seller Schedule, (ii) as consented to in writing by Purchaser, (iii) as required by applicable Law or (iv) otherwise contemplated by the terms of this Agreement or any Ancillary Agreement, from the Execution Date to the Closing Date or the earlier termination of this Agreement in accordance with Article VIII (the “Pre-Closing Period”), the Seller shall, and shall cause the Selling Affiliates to:
(i)conduct the Business in the ordinary course in a manner consistent with past practice (provided, that no action taken or not taken by the Seller or any Selling Affiliate in order to comply with clause (v) below shall be deemed a breach of this clause (i));
(ii)use commercially reasonable efforts to (A) preserve substantially intact its present business operations and organization in each case relating to the Business, (B) retain the services of the Business Employees set forth on Section 1.1(a) of the Seller Schedule and (C) preserve the goodwill of its present relationships with Persons having material business dealings with Seller (including customers and suppliers) in connection with the Business;
(iii)maintain (A) the Purchased Assets in their current condition, ordinary wear and tear, casualty and condemnation excepted, and (B) insurance upon all of the Purchased Assets in such amounts and of such kinds comparable to that in effect on the Execution Date;
(iv)pay all maintenance and similar fees and take all appropriate actions necessary to prevent the abandonment, loss or impairment of all Purchased Intellectual Property; and
(v)not take any of the following actions with respect to the Business:
35
(A)sell, transfer, lease, license, permit to lapse or otherwise dispose of any material Purchased Assets except, in each case, for sale of inventory in the ordinary course of business consistent with past practice;
(B)mortgage, pledge or otherwise encumber any Purchased Assets, except in the ordinary course of business consistent with past practice or for mortgages, pledges or encumbrances which will be released at or prior to the Closing;
(C)terminate any Material Contract (other than at the end of the term of such Material Contract in accordance with its terms), make any material amendment or modification to or waive any material right under any Material Contract, or enter into any Contract that, if entered into prior to the date hereof, would constitute a Material Contract other than customer contracts entered into in the ordinary course of business relating to the sale of Products;
(D)enter into any agreement or arrangement which would limit or restrict the distribution or sale of any Subject Product in any material respect following the Closing;
(E)accelerate the delivery or sale of the Product or any services provided by Seller or any Seller Affiliate in respect of the Business, or offer discounts, rebates or special promotions on the Product products or such services that have the effect of accelerating sales to customers or delay the payment of suppliers beyond the respective payment date, in each case, to the extent such activities would be outside the ordinary course of business consistent with past practice;
(F)except as required by applicable Law or any Business Benefit Plan as in existence on the date hereof and made available to Purchaser prior to the date hereof (1) grant any loan to, or increase the compensation or benefits payable or to become payable to any Business Employee, other than increases to base salary or wage rate in the ordinary course of business consistent with past practice, of the applicable Business Employee’s base salary or annualized wage rate; (2) grant or increase any severance or termination pay for any Business Employee; (3) pay or award, or commit to pay or award, any bonuses, retention or incentive compensation to any Business Employee other than (i) payments in the ordinary course of business pursuant to past practice based on actual performance for completed performance periods, (ii) the bonuses contemplated by Section 6.11(e) and (iii) as set forth on Section 6.1(a)(v) of the Seller Schedule; (4) take any action to accelerate the time of payment, vesting or funding of any compensation or benefits to any Business Employee or (5) except as set forth on Section 6.1(a)(v) of the Seller Schedule, establish, adopt, enter into, amend, modify or terminate any Business Benefit Plan with respect to any Business Employee, except for any broad-based amendments or modifications to any Business Benefit Plan made in the ordinary course of business consistent with past practice that apply to employees of the Seller and Seller Affiliates generally;
(G)(1) terminate (without cause), furlough or layoff (including temporarily) any Business Employee; (2) except as set forth on Section 6.1(a)(v) of the
36
Seller Schedule, hire or transfer any employee to a sales, marketing, quality assurance, manufacturing or regulatory affairs role that is primarily or exclusively dedicated to the Business; (3) materially change the role or responsibilities of any Business Employee, including the percentage of time such Business Employee spends dedicated to the Business;
(H)take any action or make any omission that may undermine, restrict or limit in any way the validity or scope of any Purchased Regulatory Approval;
(I)grant to any Person any license, sublicense, covenant not to sue, immunity authorization, release or other right with respect to any Purchased Intellectual Property, or assign or transfer to any Person any rights to any Purchased Intellectual Property;
(J)Commence any Action relating to the Product or the Business, except with respect to matters arising under or in connection with this Agreement or the Ancillary Agreements;
(K)adopt a plan or agreement of complete or partial liquidation or dissolution; or
(L)agree or commit to do any of the foregoing.
(b)During the Pre-Closing Period, Seller shall provide Purchaser with written notice as soon as practicable upon any of the following, if related to the Product: (i) the initiation or receipt of any Notice and any adverse inspectional findings or Notices, in each case alleging material non-compliance with any Law (including Regulatory Laws); (ii) any material adverse audit findings or complaints; (iii) any material quality, safety, efficacy, or performance issues or corrective, preventive, or remedial actions, including recalls; and (iv) its knowledge of any Action pending or threatened against the Seller or any of the Selling Affiliates or their respective assets.
(c)During the Pre-Closing Period and for a period ending six months after the Closing, Seller shall provide Purchaser with prompt written notice upon receipt by it or any of its Affiliates of notice of an Abbreviated New Drug Application (ANDA) filing of a Generic Version of the Product.
(d)Nothing contained in this Agreement is intended to give Purchaser or its Affiliates, directly or indirectly, the right to control or direct the Business prior to the Closing. Prior to the Closing, the Seller and its Affiliates shall exercise, consistent with the terms and conditions of this Agreement, complete control and supervision over their businesses and operations.
Section 6.2Access to Information.
(a)During the Pre-Closing Period, the Seller shall, and shall cause the Selling Affiliates to, afford Purchaser and its accountants, counsel and other authorized representatives reasonable access, upon reasonable prior notice during normal business hours, to the properties,
37
books and records of the Seller and the Selling Affiliates to the extent exclusively relating to the Business or the Purchased Assets; provided, however, that such access does not interfere or disrupt the normal operations of the Seller or any of the Selling Affiliates; provided, further, however, that the Seller and the Selling Affiliates shall not be obligated to provide such information if doing so would, (i) based on advice of the Seller’s counsel, (A) create any potential Liability under applicable Laws, (B) jeopardize the protection of any attorney-client or other legal privilege, or (C) result in the improper disclosure of any trade secrets of third parties or (ii) in the reasonable judgment of the Seller, violate a Contract or obligation of confidentiality owing to a third party. All requests for information made pursuant to this Section 6.2(a) shall be directed to such person or persons as may be designated by the Seller, and Purchaser shall not directly or indirectly contact any officer, director, employee, agent or representative of the Seller or any of its Affiliates without the prior approval of such designated person(s). Purchaser acknowledges and agrees that any information provided to Purchaser or its representatives pursuant to this Section 6.2(a) or otherwise by or on behalf of the Seller, its Affiliates or their representatives shall be subject to the terms and conditions of Section 6.3.
(b)After the Closing Date, Purchaser shall grant to Seller and its Affiliates such access to financial records and other information in its possession related to Purchaser’s conduct of the Business and such cooperation and assistance as shall be reasonably required to enable Seller and its Affiliates to complete their legal, regulatory, stock exchange and financial reporting requirements and for any other reasonable business purpose, including in respect of litigation and insurance matters; provided, however, that such access does not unduly interfere or disrupt the normal operations of Purchaser or any of its Affiliates; provided, further, that Purchaser shall not be obligated to provide such information if doing so would (i) based on advice of Purchaser’s counsel, create any potential Liability under applicable Laws or would jeopardize the protection of any attorney-client or other legal privilege, or (ii) in the reasonable judgment of Purchaser, would (A) result in the improper disclosure of any trade secrets of third parties or (B) violate a Contract or obligation of confidentiality owing to a third party. The Seller shall promptly reimburse Purchaser for Purchaser’s reasonable out-of-pocket expenses associated with requests made by the Seller and its Affiliates under this Section 6.2(b), but no other charges shall be payable by Seller to Purchaser in connection with such requests; provided, further, that this Section 6.2(b) shall not apply with respect to access required for Tax matters, which are the subject of Article IX.
(c)For a period of seven years following the Closing Date, Purchaser shall, and shall cause its controlled Affiliates to, preserve and retain all corporate, accounting, legal, auditing, human resources and other books and records of the Business relating to the conduct and operations of the Business prior to the Closing Date and shall not destroy, alter or otherwise dispose of any such books and records of the Business without first giving reasonable prior notice to the Seller and offering to surrender to the Seller such books and records or such portions thereof, to the extent practicable and legally permissible.
(d)Purchaser acknowledges and agrees that prior to making any records available to Purchaser, the Seller and its Affiliates may redact any portions thereof that do not relate to the Business, the Product, the Purchased Assets or the Assumed Liabilities.
38
(e)Seller shall prepare one or more flash drives (or similar storage medium) containing electronic copies of the Data Room as of the date of this Agreement (but at least three (3) hours prior to the execution thereof) and deliver such flash drives (or similar storage medium) to Purchaser prior to the Closing.
Section 6.3Confidentiality.
(a)Prior to the Closing, Seller and Purchaser acknowledge that they each are and remain bound by the terms of that certain Mutual Confidential Disclosure Agreement (as amended, the “MCDA”), dated as of October 26, 2021, by and between Seller and Purchaser. Each of Seller and Purchaser shall abide by the terms of the MCDA, the terms of which are incorporated herein by reference. The MCDA will remain in full force and effect until the Closing in accordance with Article VIII and thereafter all Confidential Information of Seller relating exclusively to the Purchased Assets as of the Closing Date shall constitute Confidential Information of Purchaser. For clarity, Seller shall retain all Confidential Information of Seller relating to the Retained Liabilities.
(b)Subject to Section 6.3(a), for a period of six years following the Closing, each of Purchaser and Seller agree that they and each of their respective controlled Affiliates and representatives shall keep confidential and safeguard all, and not use any, Confidential Information concerning the other Party (which, in the case of Purchaser, shall include information concerning the CVC Network), as applicable, or any of their business operations. Each of Purchaser and Seller shall be liable for any unauthorized disclosure of Confidential Information or other violation of the obligations set forth in this Section 6.3(b) by any of its controlled Affiliates or representatives as if it were their own failure to comply with such obligations.
(c)Notwithstanding anything to the contrary in this Section 6.3 or the MCDA and subject to the requirements to maintain and preserve Privileges in accordance with Section 6.10, in the event that any of Purchaser, the Seller or any of their respective Affiliates is required by applicable Law or a Governmental Authority to disclose any of the Confidential Information that such Person is required to keep confidential pursuant to Section 6.3(a) or (b), such required disclosure shall be permitted so long as, the Party with such confidentiality obligation shall (i) to the extent legally permissible, prior to any such disclosure, (A) provide the other Party with prompt notice of such request or requirement describing the circumstance of such request or requirement and the scope of information which shall be made available and (B) reasonably consult with the other Party with a view to avoiding the disclosure or narrowing its scope, including if requested, taking all reasonable steps to resist or avoid any such disclosure and (ii) if compliance with the obligations set forth in the preceding clause (i) shall not be legally permissible, as soon as reasonably practicable and legally permissible after the disclosure, inform the other party of the circumstances of the requirement to disclose Confidential Information and about the disclosure which was made.
Section 6.4Efforts; Antitrust Notifications.
(a)Purchaser and Seller shall, and shall cause their respective controlled Affiliates to, (i) use reasonable best efforts to promptly obtain all authorizations, consents, orders
39
and approvals of all Governmental Authorities and officials that may be or become necessary or advisable for its execution and delivery of, and the performance of its obligations pursuant to, this Agreement and the Ancillary Agreements, (ii) cooperate fully with the other Party in promptly seeking to obtain all such authorizations, consents, orders and approvals, and (iii) provide such other information to any Governmental Authority as such Governmental Authority may request in connection herewith. Each Party hereto, as applicable, agrees to, and to cause its Affiliates to, use its reasonable best efforts to obtain expiration or an early termination of the applicable waiting period, including under the HSR Act, and to supply as promptly as practicable to the appropriate Governmental Authorities any additional information and documentary material that may be requested pursuant to the HSR Act. Neither the Seller, on the one hand, nor Purchaser, on the other hand, may (or may permit any of their respective Affiliates to), without the consent of the other Party, (A) cause any such filing or submission applicable to it to be withdrawn or refiled for any reason, including to provide the applicable Governmental Authority with additional time to review any of the transactions contemplated by this Agreement, or (B) consent to any voluntary extension of any statutory deadline or waiting period or to any voluntary delay of the consummation of the transactions contemplated by this Agreement at the behest of any Governmental Authority. Purchaser and Seller shall each be responsible for paying fifty percent (50%) of all fees and make all other payments to any Governmental Authority in order to obtain any such authorizations, consents, orders or approvals. Notwithstanding anything to the contrary in this Agreement, nothing in this Agreement shall require Purchaser or any of its Affiliates or any other Person to (i) propose, negotiate, commit to and effect, by consent decree, hold separate or asset preservation orders or otherwise, the sale, divesture, disposition, or license of any companies, assets, operations, properties, produce, rights, licenses, services or businesses of Purchaser or any of its Affiliates or any other Person; (ii) contest, resist, or resolve any Action that is commenced or threatened by a Governmental Authority or other Person challenging the transactions contemplated by this Agreement or the Ancillary Agreements under antitrust Law or to have vacated, lifted, reversed or overturned any decree, judgment, injunction or other order, whether temporary, preliminary or permanent, that is in effect and that prohibits, prevents or restricts consummation of the transactions contemplated by this Agreement, or to oppose any such Actions, whether judicial or administrative against it in connection with transactions contemplated by this Agreement or the Ancillary Agreements; (iii) agree to any conditions relating to, or changes or restrictions in, the operations of any such assets, businesses or interests; or (iv) agree to any material modification or waiver of the terms and conditions of this Agreement or the Ancillary Agreements.
(b)To the extent permitted by applicable Law or the applicable Governmental Authority, each of Purchaser, on the one hand, and the Seller, on the other hand, shall promptly notify the other of any communication it or any of its Affiliates receives from any Governmental Authority, relating to the matters that are the subject of this Agreement and permit the other to review in advance any proposed communication by such Party to any Governmental Authority and, to the extent reasonably practicable, each will consult with the other on and consider in good faith the views of the other in connection with, all of the information relating to Purchaser or the Seller, as the case may be, and any of their respective Affiliates, that appears in any filing made with, or written materials submitted to, any third party or any Governmental Authority in connection with this Agreement and the other transactions contemplated by this Agreement. Unless required by a Governmental Authority, neither Purchaser, on the one hand, nor the Seller, on the other hand, shall (or permit any of their respective Affiliates to) agree to participate in any
40
communication with such Governmental Authority in respect of any filings, investigation (including any settlement of the investigation), litigation or other inquiry unless it consults with the other in advance and, to the extent permitted by such Governmental Authority, gives the other the opportunity to attend and participate in such communication. Purchaser, on the one hand, and the Seller, on the other hand, will, and will cause their respective Affiliates to, coordinate and cooperate fully with each other in exchanging such information and providing such assistance as the other may reasonably request in connection with the foregoing and in seeking early termination of any applicable waiting periods, including under the HSR Act. Purchaser, on the one hand, and the Seller, on the other hand, will promptly provide each other with copies of all correspondence, filings or communications between them or any of their representatives or Affiliates, on the one hand, and any Governmental Authority or members of its staff, on the other hand, with respect to this Agreement and the transactions contemplated by this Agreement; provided, however, that such materials may be redacted (i) to remove references or materials concerning the valuation of the Business or other material reasonably determined by a Party to be commercially sensitive, (ii) as necessary to comply with contractual arrangements, and (iii) as necessary to address reasonable attorney-client or other privilege or confidentiality concerns, to the extent that such attorney-client or other privilege or confidentiality concerns are not governed by a common interest privilege or doctrine.
(c)Purchaser, on the one hand, and the Seller, on the other hand, shall not enter into any transaction, or any Contract or other written agreement, to effect any transaction (including any merger or acquisition) that might reasonably be expected to and has the effect of making it more difficult to: (i) obtain the expiration or termination of the waiting period under the HSR Act and approval under the Foreign Merger Control Laws; (ii) avoid the entry of, the commencement of litigation seeking the entry of, or to effect the dissolution of, any injunction, temporary restraining order or other order that would prevent the consummation of the transactions contemplated by this Agreement; or (iii) obtain all authorizations, consents, orders and approvals of Governmental Authorities that may be or become necessary or advisable for the consummation of the transactions contemplated by this Agreement; provided, for the avoidance of doubt, that nothing in this Section 6.4(c) shall limit any members of the CVC Network from entering into any such transactions, Contracts or other written agreements.
Section 6.5Services from Affiliates. Purchaser acknowledges that the Business currently receives from the Seller and its Affiliates (including through Contracts to which the Seller or its Affiliates are party that are Excluded Assets) certain administrative, corporate and other services and benefits, including accounting, legal, intellectual property related services, information technology services reporting and back office services and processing, financial systems, treasury services (including insurance, human resources, cash management, financing, taxation and internal audit), research and development (including pharmacovigilance and operations), regulatory and medical affairs, technical development, quality, product supply (including procurement, production, CMO management, distribution and quality), product safety, environmental management and marketing and sales support. Other than as may be provided pursuant to the terms of the Transitional Services Agreement, Purchaser further acknowledges that all such services and benefits shall cease, and any agreement in respect thereof shall terminate with respect to the Business as of the Closing Date and thereafter the Seller’s and its Affiliates’ sole obligation with respect to the provision of any services with respect to the Business shall be as set forth in the Transitional Services Agreement.
41
Section 6.6Publicity. Neither Purchaser, on the one hand, nor the Seller, on the other hand, will issue or permit any of their respective controlled Affiliates to issue any press release, website posting or other public announcement with respect to this Agreement or the transactions contemplated hereby (other than the Debt Financing) without the prior consent of the other Party (such consent not to be unreasonably withheld, conditioned or delayed), except as may be required by Law or stock exchange rules or regulations (in which case the parties will endeavor to allow the other Party to comment on such release or statement to the extent practicable); provided, however, that subject to Section 6.11(g), Purchaser, on the one hand, and the Seller, on the other hand, may make internal announcements to their respective employees that are consistent with the Parties’ prior public disclosures regarding the transactions contemplated by this Agreement and the Ancillary Agreements and provided, further, that members of the CVC Network may provide general information about the subject matter of this Agreement, the Ancillary Agreements and the Business (including its performance and improvements) in connection with such members’ fund raising, marketing, informational or reporting activities in the ordinary course. If either of the Seller or Purchaser, based on the advice of its counsel, determines that this Agreement, or any of the other Ancillary Agreements, must be publicly filed with a Governmental Authority, then the Seller or Purchaser, as applicable, prior to making any such filing, shall, subject to applicable Law, provide the other Party and its counsel with a redacted version of this Agreement (and any other Ancillary Agreement) which it intends to file, and will give due consideration to any comments provided by the other Party or its counsel and use commercially reasonable efforts to ensure the confidential treatment by such Governmental Authority of those sections specified by the other Party or its counsel for redaction and confidentiality. At any time following the issuance of the initial press release, any Party, its Affiliates and representatives, shall be permitted to make any public announcements regarding this Agreement, the Ancillary Agreements, the Acquisition and the other transactions contemplated hereby and thereby without the prior written consent of any other Parties, to the extent such announcements are consistent with such press release or other prior disclosures approved in accordance with this Section 6.6. Notwithstanding any other provision of this Agreement, the requirements of this Section 6.6 shall not apply to any disclosure of the Seller or Purchaser of any information concerning this Agreement, the Ancillary Agreements or the transactions contemplated hereby or thereby in connection with any dispute between the Parties regarding this Agreement, the Ancillary Agreements, or the transactions contemplated hereby or thereby. For the avoidance of doubt, nothing herein shall restrict (i) Purchaser, the Lenders or their respective Affiliates from making customary announcements and communications in connection with the arrangement and syndication of the Debt Financing or (ii) Purchaser or its Affiliates from making customary announcements and communications in connection with their normal fund raising, marketing, informational or reporting activities. Purchaser and Seller will agree on the language of press releases at the execution of this Agreement and the closing of the Acquisition.
Section 6.7Use of Seller Names and Marks.
(a)Purchaser hereby acknowledges that the Seller and its Affiliates own all right, title and interest in and to the Seller Names and Marks, together with all variations and acronyms thereof and all Trademarks or goodwill containing, incorporating, based on or associated with any of the foregoing, and that, except as may be expressly provided in the Transitional Trademark License Agreement, any and all right of the Business to use the Seller
42
Names and Marks shall terminate as of the Closing and shall immediately revert to the Seller and its Affiliates along with any and all goodwill associated therewith. Purchaser further acknowledges that it has no rights or interests, and is not acquiring any rights or interests, directly or indirectly, through the Business or otherwise, to use the Seller Names and Marks, except as may be expressly provided in the Transitional Trademark License Agreement.
(b)Except as may be expressly provided in the Transitional Trademark License Agreement, no right to use the Seller Names and Marks is granted by the Seller or any of its Affiliates to Purchaser or its Affiliates, whether by implication or otherwise, and nothing hereunder permits Purchaser or its Affiliates to use the Seller Names and Marks in any manner or to register or seek to register, or to permit, cause or assist any third party to register or to seek to register, any of the Seller Names and Marks in any jurisdiction.
Section 6.8Further Action. On the terms and subject to the conditions of this Agreement (including Section 6.4), each Party shall use its commercially reasonable efforts (except to the extent a higher standard is provided for herein, in which case, the applicable Party shall use efforts that meet such higher standard) to take or cause to be taken in an expeditious manner all actions and to do or cause to be done all things necessary or appropriate to satisfy the conditions to the Closing, to consummate the transactions contemplated hereby and by the Ancillary Agreements and to comply promptly with all legal requirements that may be imposed on it or any of its Affiliates with respect to the Closing. Subject to appropriate confidentiality protections, each of the Parties will cooperate with and furnish to the other Party such necessary information and reasonable assistance as such other Party may reasonably request in connection with the foregoing.
Section 6.9Bulk Sales Law. Purchaser acknowledges that the Seller and its Affiliates have not taken, and do not intend to take, any action required to comply with any applicable bulk sale or bulk transfer Laws or similar Laws of any jurisdiction. Purchaser hereby waives compliance by the Seller and its Affiliates with the provisions of all applicable so called “bulk sales” or “bulk transfer” laws in connection with the transactions contemplated by this Agreement.
Section 6.10Litigation; Privileged Matters.
(a)Purchaser agrees to, after the Closing, cooperate with the Seller and its Affiliates as necessary in connection with the defense by the Seller or any of its Affiliates against, or the prosecution of, any Action or investigation, whether existing, threatened, or anticipated, relating to any Subject Product, the Business or the Purchased Assets, including (i) the ownership of the Purchased Assets prior to the Closing, (ii) the research, development, marketing, importation, sale or other Exploitation of the Product prior to the Closing, (iii) the Manufacture or production of the Product sold prior to the Closing, (iv) the Purchased Contracts with respect to any period prior to the Closing and (v) the operation or conduct of the Business prior to the Closing. In each such case as described in the foregoing sentence, Purchaser’s cooperation shall (1) be in addition to Purchaser’s obligation to indemnify the Seller Indemnitees for the Assumed Liabilities associated with any such Action and (2) include providing the access described in Section 6.2(b).
43
(b)The Parties agree that, from and after the Closing, to the extent permitted by Law, their respective rights and obligations to maintain, preserve, assert or waive any or all attorney-client and work product privileges belonging to any Party with respect to the Business and the Purchased Assets, the Excluded Assets, the Assumed Liabilities and the Retained Liabilities (collectively, “Privileges”) shall be governed by the provisions of this Section 6.10. From and after the Closing, with respect to (i) matters relating to the Excluded Assets or the Retained Liabilities, (ii) all information relating to the Acquisition (other than with respect to information solely possessed by Purchaser), and (iii) matters relating to the Business prior to the Closing, the Seller and its Affiliates shall have sole authority in perpetuity to determine whether to assert or waive any or all Privileges, and Purchaser and its Affiliates shall take no action without the prior written consent of the Seller that would be reasonably likely to result in any waiver of any such Privilege that could be asserted by the Seller or any of its Affiliates. From and after the Closing, with respect to (1) matters relating to the Business on or after the Closing, (2) Purchased Assets or (3) the Assumed Liabilities (in each case, for the avoidance of doubt, excluding all information relating to the sale of the Business (other than with respect to information solely possessed by Purchaser)), Purchaser shall have sole authority in perpetuity to determine whether to assert or waive any or all Privileges, and the Seller and the Selling Affiliates shall take no action without the prior written consent of Purchaser that would be reasonably likely to result in any waiver of any such Privilege. All information as to which Seller or any of its Affiliates, on the one hand, or Purchaser or any of its Affiliates, on the other hand, would be entitled to assert or has asserted a Privilege pursuant to this Section 6.10 is referred to as “Privileged Information”.
(c)From and after the Closing, upon (i) receipt by the Seller or any of the Selling Affiliates, on the one hand, or Purchaser or any of its Affiliates, on the other hand, of any subpoena, discovery or other request from any third party that actually or arguably calls for the production or disclosure of Privileged Information of the other or (ii) the Seller or the Selling Affiliates, on the one hand, or Purchaser or any of its Affiliates, on the other hand, obtaining knowledge that any current or former employee of the other has received any subpoena, discovery or other request from any third party that actually or arguably calls for the production or disclosure of Privileged Information of the other Party, the Seller or Purchaser, as the case may be, shall promptly notify in writing the other Party of the existence of the applicable request and shall provide the other a reasonable opportunity to review such request and to assert any rights it may have under this Section 6.10 or otherwise to prevent the production or disclosure of Privileged Information, except that if any such request relates to Privileged Information relating to the Acquisition (other than with respect to information solely possessed by Purchaser), the Seller and the Selling Affiliates shall have no obligation under this Section 6.10(c) to notify Purchaser or provide Purchaser any opportunity to review such request.
Section 6.11Employee Matters.
(a)Acquired Employees. From and after the date of this Agreement until the Closing Date, Seller shall deliver to Purchaser, on a periodic basis as reasonably requested by Purchaser, an updated Business Employee List, including the information described in Section 3.10(f) with respect to each Business Employee. No later than 10 days prior to the Closing Date, Purchaser (or an Affiliate of Purchaser) shall, in writing, offer employment to all Business Employees. The offered employment must (i) not require the Business Employee to work from a
44
principal place of employment that is more than 35 miles from the Business Employee’s assigned office location immediately prior to Closing and (ii) be at a base salary or wage at least equal to the Business Employee’s base salary or wage on the Closing Date (the “Required Terms”). Any Business Employee who is offered employment by Purchaser (or Purchaser’s Affiliate) and who accepts such offer of employment shall commence employment with Purchaser (or Purchaser’s Affiliate) as of and contingent upon the Closing (such employee who commences employment with Purchaser, an “Acquired Employee”). All Acquired Employees shall be employed solely on an “at will” basis, except as required by applicable Law. As of and contingent upon the Closing, the Seller (or Seller’s applicable Affiliate) shall terminate the employment (or enter into a mutual termination agreement) of (or with) each Acquired Employee. Any Business Employee who is not actively at work on the Closing Date due to an approved leave of absence or to a short-term disability (an “Inactive Employee”) shall be offered employment with Purchaser (or Purchaser’s Affiliate) on the Required Terms when and if such Business Employee presents himself or herself to Purchaser (or Purchaser’s Affiliate) for active employment during the six-month period beginning on the Closing Date (or such longer period that may be required under applicable Law). Any such Inactive Employee who accepts employment with Purchaser (or Purchaser’s Affiliate) shall be treated as an Acquired Employee under this Section 6.11 beginning on the individual’s date of hire with Purchaser (or Purchaser’s Affiliate). Any Inactive Employee who does not present himself or herself for active employment with Purchaser (or Purchaser’s Affiliate) within the six-month period beginning on the Closing Date (or such longer period that may be required under applicable law) shall not be offered continued employment with Purchaser (or Purchaser’s Affiliate) and, notwithstanding anything in this Agreement to the contrary, Purchaser shall not assume any liabilities in respect of such Inactive Employee.
(b)Compensation and Benefits. For the [***]-year period immediately following the Closing Date (or if earlier, until the date Separate Payment is no longer in effect), Purchaser (or Purchaser’s Affiliate) shall provide the Acquired Employees with (i) base salary or wage at least equal to the Business Employee’s base salary or wage on the Closing Date, and (ii) other compensation and benefits (excluding equity and equity-based awards, non-qualified deferred compensation, severance benefits, defined benefit pension plans, post-termination and retiree health and welfare benefits, retention and transaction based payments) that are in the aggregate substantially similar to the other compensation and benefits offered to the Acquired Employees by the Seller or its applicable Affiliate immediately prior to Closing.
(c)Employment Guarantee Period. If Purchaser or any of Purchaser’s controlled Affiliates terminates the employment of any Acquired Employee other than for cause or due to the fact that Separate Payment is no longer in effect during the [***]-year period immediately following the Closing Date (“Employment Guarantee Period”), Purchaser or Purchaser’s applicable Affiliate will pay to the Acquired Employee a severance benefit on the terms set forth in Section 6.11(c) of the Seller Schedule. Purchaser and Purchaser’s Affiliates shall not have any obligations under this Section 6.11(c), (1) if the employee is terminated for cause or due to the fact that Separate Payment is no longer in effect or (2) in the event an employee voluntarily terminates employment. Purchaser shall be permitted to condition the benefits under this paragraph on the Acquired Employee entering a customary release of all claims and such release becoming effective in accordance with its terms where permitted by applicable Law.
* [***] CERTAIN IDENTIFIED INFORMATION HAS BEEN EXCLUDED FROM THE EXHIBIT BECAUSE IT BOTH (A) IS NOT MATERIAL AND (B) WOULD BE COMPETITIVELY HARMFUL IF PUBLICLY DISCLOSED
45
(d)Benefit Plans. All Acquired Employees shall be eligible to participate in Purchaser’s employee benefit plans pursuant to the terms and conditions of such plans. Acquired Employees shall be given credit, without duplication, for service time with the Seller or its applicable Affiliate for purposes of eligibility and vesting, including with respect to level of vacation, under Purchaser’s plans (other than with respect to equity and equity-based awards, non-qualified deferred compensation, defined benefit pension plans, post-termination and retiree health and welfare benefits, retention and transaction based payments) to the same extent and for the same purpose as such service was credited to such Acquired Employees under the analogous employee benefit plan of the Seller or its applicable Affiliate immediately prior to the Closing. During the plan year in which the Closing Date occurs, Purchaser shall, with respect to any eligible Acquired Employee or, as applicable, a family member of an eligible Acquired Employee, use commercially reasonable efforts to (i) waive any waiting period, (ii) waive any exclusion or limitation for preexisting conditions which were covered under any group health plan maintained by the Seller prior to the Closing Date, and (iii) grant credit (for purposes of annual deductibles, co-payments and out-of-pocket limits) for any covered claims incurred or payments made prior to the Closing Date.
(e)Closing-Year Bonuses. With respect to Seller bonus and commission incentives for which the applicable performance period has ended on or prior to the Closing Date, or which has otherwise been earned, and in which any Acquired Employee participates, Seller shall pay to each such Acquired Employee the bonus or commission earned based on actual performance and without respect to any continued service requirements, no later than the date on which Seller pays such bonuses or commissions to other similarly situated employees of Seller. With respect to any Seller bonus or commission incentives for which the applicable performance period has not ended on or prior to the Closing Date and in which any Acquired Employee participates, Seller shall pay to each Acquired Employee a prorated annual bonus or commission, as applicable, the amount of which shall equal the product of the applicable Acquired Employee’s annual bonus or commission (based on actual performance) multiplied by a fraction, the numerator of which is the number of days in the calendar year during which the Closing Date occurs that elapse prior to the Closing Date, and the denominator of which is 365. Such prorated bonuses and commissions shall be paid no later than the date on which Seller pays such bonuses to other similarly situated employees of Seller. Notwithstanding the above, any bonus or commission payable under this paragraph (e) shall be paid no later than June 30th of the calendar year following the year to which such bonus relates.
(f)Accrued Vacation, Sick Leave and Personal Time. Seller shall pay each Acquired Employee all accrued but unused vacation and paid time-off for periods prior to the Closing Date as soon as administratively practicable following the Closing Date.
(g)401(k) Plan. Seller shall take all actions necessary or appropriate to cause all Acquired Employees who participate in the Seller 401(k) Plan (the “Seller 401(k) Plan”) to be fully vested in their account balances under the Seller 401(k) Plan, and shall make to the Seller 401(k) Plan all employer contributions that would have been made on behalf of such employees had the transactions contemplated by this Agreement not occurred, regardless of any service or end of year employment requirements, but prorated for the portion of the plan year that ends on the Closing Date.
46
(h)Employee Communications. Any broad-based written (or prepared oral) communications proposed to be made to any Business Employees pertaining to compensation or benefit entitlements to be provided following the Closing Date or any other employment matters resulting from the transactions contemplated in this Agreement shall be subject to prior approval of Purchaser, which shall not be unreasonably withheld, conditioned or delayed. In furtherance of the foregoing, Seller shall provide Purchaser with a copy of the intended communication (including, in the case of any such oral communications, copies of scripts, talking points or other similar materials), and Seller shall have a reasonable period of time to review and comment on the communication.
(i)No Third Party Beneficiaries. Nothing in this Agreement shall create any third party beneficiary or other rights: (i) in any other Person, including any Business Employee, any participant in any employee benefit plan of Seller or Purchaser or their respective Affiliates, or to (ii) continued employment with Purchaser or any of its Affiliates or to any particular term or condition of employment. Notwithstanding anything to the contrary in this Agreement, nothing contained herein shall confer upon any Business Employee any right to continue in the employ or service of Purchaser or any of its Affiliates, or shall restrict in any manner the right of Purchaser to discharge, terminate or modify any compensation arrangement of any Acquired Employee for any reason at any time. Nothing in this Section 6.11 shall (a) be deemed or construed to be an amendment or other modification of any employee benefit plan of Purchaser or any of its Affiliates, or (b) create any third party rights in any Business Employee, or other current or former employee or service provider of Seller or its Affiliates (or any beneficiaries or dependents thereof).
(j)Non-Solicitation. From the Closing Date and for a period of one year thereafter, Purchaser shall not, and shall not permit any of its Affiliates to, directly or indirectly, hire or solicit any senior (director level or above) employee of Seller with whom Purchaser came into contact in connection with its evaluation of the Acquisition (other than the Business Employees) or encourage any such employee to leave such employment or hire any such employee who has left such employment, except (i) pursuant to a general solicitation or advertisements which is not directed specifically to any such employees, (ii) employment efforts made through employment agencies and third party recruiters and (iii) for such employees who have approached Purchaser in an unsolicited manner; provided that nothing in this Section 6.11(j) shall prevent Purchaser or any of its Affiliates from soliciting or hiring (i) any employee whose employment has been terminated by Seller; (ii) after 90 days from the date of termination of employment, any employee whose employment has been terminated by the employee or (iii) any Business Employees.
Section 6.12WARN Act. Purchaser agrees to provide any required notice under the WARN Act, and any similar Law, and to otherwise comply with the WARN Act and any such other similar Law with respect to any “plant closing” or “mass layoff” (as defined in the WARN Act) or group termination or similar event affecting Acquired Employees occurring after the Closing Date and Seller and Seller’s Affiliates agree to provide any required notice under the WARN Act, and any similar Law, and to otherwise comply with the WARN Act and any such other similar Law with respect to any “plant closing” or “mass layoff” or group termination or similar event affecting any Business Employees on or prior to the Closing Date. Seller shall
47
notify Purchaser of any “employment loss” (as defined in WARN) of any Business Employees that occur during the 90-day period prior to the Closing.
Section 6.13Accounts Payable. In the event that, subsequent to the Closing, Purchaser or an Affiliate of Purchaser receives any invoices from any third party with respect to any account payable relating to the Business outstanding prior to the Closing, then Purchaser shall, within 30 days after receipt of such invoice, provide a copy of such invoice to Seller. In the event that, subsequent to the Closing, Seller or any of its Affiliates receives any invoices from any third party with respect to any account payable relating to the Business for any period after the Closing, then Seller shall, within 30 days after receipt of such invoice, provide a copy of such invoice to Purchaser.
Section 6.14Wrong Pockets; Payments.
(a)After the Closing, if the Seller or any of its Affiliates is the owner of, receives or otherwise comes to possess any Purchased Asset, the Seller promptly shall notify Purchaser and transfer or cause to be transferred, such Purchased Asset to Purchaser. After the Closing, if Purchaser or any of its Affiliates is the owner of, receives or otherwise comes to possess any Excluded Asset or any other asset that is unrelated to the Business or properly belongs to the Seller or any of its Affiliates in accordance with the terms of this Agreement, Purchaser shall promptly notify the Seller and transfer or cause to be transferred, such Excluded Asset or other asset to the Seller (or to the Affiliate designated by the Seller). Prior to any such transfer, the Person receiving or possessing such asset shall hold such asset for the benefit of any such other Person and shall not use such asset (or payment in respect of such asset) for any other purpose.
(b)In the event that, after the Closing Date, any Party receives any payments or other funds due to the other Party pursuant to the terms of any of this Agreement or the Ancillary Agreements, then the Party receiving such funds shall promptly (and in any case within 15 Business Days) forward such funds to the proper Party. The Parties acknowledge and agree there is no right of offset regarding such payments and a Party may not withhold funds received from third parties for the account of the other Party in the event there is a dispute regarding any other issue under any of this Agreement or the Ancillary Agreements.
(c)If the Seller or any of its Affiliates makes any deposit or payment prior to the Closing under any Purchased Contract in respect of supplies of goods for the Business not received prior to the Closing, Purchaser shall reimburse to the Seller or its applicable Affiliate, within 15 Business Days following the Closing Date, an amount equal to the portion of such deposit or payment that relates to the goods to be received after the Closing by Purchaser. If any customer or other third party offsets from any amount payable to the Seller or any of its Affiliates, any amount owed from the Business to such customer or other third party which is an Assumed Liability, Purchaser shall reimburse to the Seller or its applicable Affiliate an amount equal to such offset within 15 Business Days following Purchaser’s receipt of an invoice therefor, with reasonable supporting documentation.
Section 6.15Noncompetition. Until the expiration of the Royalty Term with respect to all Royalties, Seller shall not, and shall cause its controlled Affiliates not to, whether alone or
48
jointly with another party, directly or indirectly, knowingly carry on or be engaged or concerned or interested economically or otherwise in any manner in the development, Manufacture, distribution, sale, marketing, promotion or other commercialization of any product which competes with the Subject Products anywhere in the world or own, manage, operate or control any Person engaged in such a competing business.
Section 6.16Rights of the Parties During the Term of Certain Ancillary Agreements. The Seller or its applicable Affiliate shall retain any and all rights under Contracts, all equipment that would be considered Purchased Equipment, supplies and other fixed assets, all Regulatory Approvals and any other assets that would otherwise be considered Purchased Assets pursuant to Section 2.1 (the “Required Seller Assets”), to the extent that the Seller or such Affiliate needs such Required Seller Assets in order to provide the services or perform its obligations described in the Transitional Services Agreement. Each Required Seller Asset shall constitute Excluded Assets until the earlier of (a) such time as a Required Seller Asset is no longer necessary to perform the obligations of the Seller or of any of its Affiliates under the Transitional Services Agreement and (b) the applicable Ancillary Agreement pursuant to which the Seller or its applicable Affiliate needed to use such Required Seller Assets is terminated, at such time the Required Seller Assets will become Purchased Assets and the Parties will arrange for their transfer consistent with Article II and the Transitional Services Agreement. For the purposes of the representations and warranties of the Seller, the Parties shall deem the Required Seller Assets as having been transferred in its entirety at Closing.
Section 6.17Quality Control.
(a)Purchaser understands and agrees that, except as otherwise required under applicable Law (or as set forth under any Purchased Contract or Ancillary Agreement), from and after the Closing Date, Purchaser shall be responsible for ensuring the Subject Products are manufactured in compliance with applicable Laws, including GMP regulations applicable to the Subject Products and all other quality and manufacturing standards imposed by any applicable Governmental Authorities. From and after the Closing Date, Purchaser shall be responsible for all quality issues related to Subject Products; provided that the Seller shall, upon Purchaser’s reasonable request, provide commercially reasonable assistance, pursuant to the Seller’s standard operating procedures, to Purchaser in handling any such quality issues existing and open prior to the Closing Date related to Subject Products Manufactured, distributed or sold prior to, on or after the Closing Date, whether or not known to Seller as of the Closing Date. Purchaser shall provide any such requests in writing and provide the Seller with a reasonable time period to respond and prepare assistance. Any assistance provided by the Seller to Purchaser in handling any such quality issues shall not exceed the Seller’s current standard operating procedures in handling the quality issues as provided in the Seller’s policies and procedures that are effective at the time of the request by Purchaser.
(b)From and after the Closing, in the event that any Governmental Authority shall allege or prove that any Subject Product does not comply with any applicable Law relating to the quality and manufacturing of the Subject Products or Product Specifications, Purchaser shall be fully responsible for such investigation and the disposition thereof. Prior to the transfer of the Purchased Regulatory Approvals to Purchaser, Seller and Purchaser agree to cooperate as necessary including to take responsive action, respond to, or engage with the Governmental
49
Authority. The Party which sold the applicable Subject Product shall be responsible for all costs and expenses with respect to any such investigation and disposition of such Subject Product. The Parties shall cooperate and work together in good faith in addressing all such non-compliance allegations and occurrences.
(c)Each Party shall notify the other as soon as practicable after it becomes aware of any misbranding, adulteration, malfunction of or other defect in any Subject Product (a “Defective Product”). If a Governmental Authority issues a warning letter or threatens or commences an Action (including seeking an injunction) in relation to, seizes, or requests or requires a recall of any Subject Product, Purchaser or the Seller, as the case may be, shall immediately notify the other Party of the action, seizure, request or requirement and provide to the other Party a copy of any warning letter or notice given by the Governmental Authority. If an action as described in the foregoing sentence requires response, Purchaser, after consultation with the Seller, will determine the nature, content and scope of that response and will determine the procedures and steps in respect of that response, whether or not the response is to be given by Purchaser or the Seller.
(d)From and after Closing, Purchaser shall have the right to decide whether to undertake a recall of any Subject Product voluntarily, and the nature, level and scope of, and all steps and procedures with respect to, any such voluntary recall and if Purchaser decides to recall a Defective Product, then (i) Purchaser shall take all reasonable steps to effect the recall and (ii) Purchaser and the Seller shall use reasonable efforts to mitigate the costs of such recall.
(e)If a Governmental Authority orders the recall of any Subject Product after the Purchased Regulatory Approvals have been transferred to the Purchaser, Purchaser shall comply with any notice given by the Governmental Authority with respect to such recall. If a Governmental Authority orders the recall of any Subject Product from and after the Closing Date but prior to the date of transfer of the Purchased Regulatory Approvals to Purchaser, Purchaser shall cooperate with Seller with respect to such recall. The Party that sold the applicable Subject Product shall be responsible for all costs and expenses with respect to any recalls of any Subject Product from and after the Closing Date.
(f)Notwithstanding the foregoing, if any Defective Product that is subject to a recall (either initiated by Purchaser or ordered by a Governmental Authority) bears any of the Seller Names and Marks on the product label, Purchaser shall (i) notify the Seller of the recall decision, as soon as reasonably practicable, but in no event later than two Business Days after Purchaser decides to or is ordered to conduct the recall of the Defective Product, and (ii) provide the Seller and its applicable Affiliates with an opportunity to review any proposed materials related to the recall prior to the filing or publication thereof and any related process with respect thereto and any proposed correspondence or other communication with any Governmental Authorities with respect to any proposed or submitted recall materials.
(g)In connection with the undertakings set forth in Section 6.17(a)-(f), Purchaser agrees to reimburse the Seller for the value of any and all out-of-pocket costs and expenses incurred by the Seller or any of its Affiliates in connection with any activities undertaken by the Seller or any of its Affiliates to handle such quality issues at the request of Purchaser or as otherwise required by any Governmental Authority or applicable Law at any
50
time on and after the Closing Date to the extent related to Product Manufactured, distributed or sold on or after the Closing Date.
(h)Notwithstanding any term or condition of Section 2.7, in the event that one or more units of Subject Product that were sold by or on behalf of Seller on or prior to December 31, 2021, are recalled after December 31, 2021, then any amounts of Net Revenue or Sublicense Revenue received by Purchaser, its Affiliates or its or their assignees or licensees (or sublicensees, regardless of tier) in connection with the sale of units of Subject Product to replace such recalled units of Subject Product, shall be payable in their entirety to Seller pursuant to the same reporting and payment procedures set forth in Section 2.7 with respect to the payment of Royalties.
Section 6.18Insurance. Purchaser is aware that the Business and the Purchased Assets are currently covered by group umbrella insurance policies taken out by Seller. The coverage under such group umbrella insurance policies is only for the benefit of the Seller and its Affiliates, and not for the benefit of Purchaser or the Business and at all times after the Closing, neither the Business nor any claim made in respect of the Business will be covered by such group umbrella insurance policies. As of the Closing Date and consistent with Article II, Purchaser agrees to arrange for its own insurance policies with respect to the Purchased Assets and the Business covering all periods and agrees not to seek, through any means, to benefit from the Seller’s or any of its Affiliates’ insurance policies which may provide coverage for claims relating in any way to the Purchased Assets and the Business (including for any events, occurrences or accidents occurring on or before the Closing Date). Notwithstanding anything to the contrary hereunder, until September 30, 2022, Seller shall maintain a “run-off” product liability insurance policy upon the Product in such amounts and of such kinds comparable to that in effect on the Execution Date.
Section 6.19Trade Notification. From the Execution Date through the Closing, the Seller and Purchaser shall cooperate in good faith to agree in writing on the method and content of the notifications to customers and suppliers of the sale of the Purchased Assets to Purchaser hereunder; provided, that the Seller shall have the sole right to deliver such notifications to customers prior to the Closing. Purchaser (prior to the Closing) shall not make any other communications or give any other notices to customers or suppliers relating to the transactions contemplated hereby prior to the date of, or inconsistent with the terms of, such written agreement.
Section 6.20Transfer of Purchased Regulatory Approvals and Compliance with Regulatory Requirements.
(a)Immediately following the Closing, Purchaser shall assume all regulatory responsibilities for compliance with the requirements of the FDA and similar state and foreign Governmental Authorities relating to the Exploitation of any Subject Product to the extent that such responsibilities can be transferred to Purchaser from Seller as of the Closing Date. In each case, such requirements include the FDCA, FDA’s implementing regulations, and all similar requirements in other jurisdictions. Purchaser and Seller agree to cooperate to execute regulatory responsibilities between the Closing Date and the transfer of the Purchased Regulatory
51
Approvals to Purchaser. For the avoidance of doubt, any responsibilities not transferred as of the Closing Date will occur upon the transfer of the Purchased Regulatory Approvals to Purchaser.
(b)With respect to the Purchased Regulatory Approvals held by the Seller or a Selling Affiliate, Seller and Purchaser shall as soon as possible after the Closing Date, and in any event in accordance with the applicable time set forth on Section 6.20(b) of the Seller Schedule undertake all actions necessary to effect the transfer of such Regulatory Approvals, including complying with the requirements under applicable Laws and requests of Governmental Authorities with respect to the transfer of such Regulatory Approvals. Within the applicable time set forth on Section 6.20(b) of the Seller Schedule, each Party, as appropriate, shall, amend, update, cancel or establish any manufacturer registrations and product listings for each of the Subject Products as needed to comply with applicable Regulatory Laws. Seller will upon Purchaser’s request provide reasonable assistance to Purchaser with respect to the transfer of such Regulatory Approvals. For clarity, Purchaser shall be responsible for all out-of-pocket costs incurred in connection with any such transfers, registrations and listings, including costs arising from procurement of certain ancillary documents, registration file transfer, document transfer, archive copying and document legalization.
(c)With respect to Purchased Regulatory Approvals for which the Seller or any Selling Affiliate continues to hold in accordance with Section 2.5, the Seller shall, and shall cause the relevant Selling Affiliates to, use reasonable efforts to maintain the Purchased Regulatory Approvals, including to continue as holder or applicant of the Purchased Regulatory Approvals, to support routine maintenance and to pursue any ongoing variations, amendments and renewals of the Purchased Regulatory Approvals, which are pending as of the Closing Date, unless otherwise mutually agreed by the Parties. Purchaser shall promptly inform the Seller (without any request of the Seller being required) of any changes in the Product, Manufacturing processes or any other items requiring submission to any Governmental Authority in order to maintain the Purchased Regulatory Approvals. Purchaser shall upon Seller’s reasonable request provide reasonable assistance to the Seller with respect to the maintenance of such Purchased Regulatory Approvals. Purchaser shall reimburse the Seller for any out-of-pocket costs and expenses incurred by the Seller or any of its Affiliates in connection with the maintenance of the Purchased Regulatory Approvals.
(d)The Seller shall, however, not be obligated to continue to own and maintain such Purchased Regulatory Approvals in any jurisdiction for longer than the time set forth next to such jurisdiction on Section 6.20(b) of the Seller Schedule except to the extent Purchaser has not yet obtained an applicable Purchased Regulatory Approval as the direct result of the Seller having failed to make any required filings for a transfer of such Purchased Regulatory Approval pursuant to this Agreement. Irrespective of whether the Purchased Regulatory Approvals have been transferred or not, from and after the Closing Date, neither the Seller nor any of its Affiliates shall be responsible for conducting any studies or testing (including non-clinical, clinical and stability studies) concerning any Subject Product, except to the extent the failure by the Seller or any of its Affiliates to conduct such study would be a violation of applicable Law (in which exceptional case the Seller shall, and shall procure that its applicable Affiliates will, at the cost of Purchaser, conduct such studies until such studies may be transferred to Purchaser).
52
Section 6.21Exclusivity. During the Pre-Closing Period, the Seller or its Affiliates shall not, and they shall not authorize or knowingly permit any of their respective representatives to, directly or indirectly, (a) solicit, initiate or induce the making, submission or announcement of, or knowingly encourage, facilitate or assist, an Acquisition Proposal, (b) furnish to any Person (other than Purchaser, its Affiliates or their respective designees) any nonpublic information relating to the Business, or afford to any Person (other than Purchaser, its Affiliates or their respective designees) access to the business, properties, assets, books, records or other non-public information, or to any personnel, of the Seller, in any such case with the intent to induce the making, submission or announcement of, or the intent to encourage, facilitate or assist, an Acquisition Proposal or any inquiries that would reasonably be expected to lead to an Acquisition Proposal, (c) participate or engage in discussions or negotiations with any Person with respect to an Acquisition Proposal, or (d) enter into any Contract relating to an Acquisition Proposal. To the extent permitted by applicable Law or confidentiality obligations, Seller shall promptly notify the Purchaser if any director, executive officer or representative of Seller becomes aware of any receipt by such Person of (i) any Acquisition Proposal, (ii) any request for information that would reasonably be expected to lead to an Acquisition Proposal, or (iii) any inquiry with respect to, or which would reasonably be expected to lead to, any Acquisition Proposal, the terms and conditions of such Acquisition Proposal, request or inquiry, and the identity of the Person or group making any such Acquisition Proposal, request or inquiry. The Seller and the Selling Affiliates shall, and shall cause their respective representatives to, immediately cease and cause to be terminated any existing discussions or negotiations with any Person (other than Purchaser, its Affiliates or their respective designees) conducted heretofore with respect to any of the foregoing.
Section 6.22Financing.
(a)Purchaser or its Affiliate, as applicable, shall use its reasonable best efforts to take, or cause to be taken, all actions and do, or cause to be done, all things necessary, proper and advisable to consummate and obtain the proceeds of the Debt Financing on or prior to the Closing Date on the terms and conditions described in the Debt Commitment Letter, including using its reasonable best efforts to:
(i)maintain in effect the Debt Commitment Letter;
(ii)negotiate and enter into all of the definitive agreements with respect to the Debt Financing (the “Definitive Agreements”) consistent with the terms and conditions contained therein (including, as necessary, the “flex” provisions contained in any related fee letter) on or prior to the Closing Date or on other terms no less favorable to the Purchaser or its Affiliate, as applicable, taken as a whole (including with respect to the conditionality thereof); and
(iii)comply with its obligations in the Debt Commitment Letter and the Definitive Agreements.
(b)In the event that all conditions contained in the Debt Commitment Letter have been satisfied (or upon such funding will be satisfied), Purchaser or its Affiliate, as applicable, shall cause the Lenders to fund the Debt Financing and to pay related fees and
53
expenses on the Closing Date. Purchaser or its Affiliate, as applicable, shall comply with its obligations, and enforce its rights, under the Debt Commitment Letter.
(c)Purchaser or its Affiliate, as applicable, shall not, without the prior written consent of Seller (not to be unreasonably withheld, conditioned or delayed):
(i)permit any material amendment, replacement, supplement or modification to, or any material waiver of any provision or remedy under, the Debt Commitment Letter if such amendment, modification, waiver or remedy could be reasonably expected to prevent, impede or delay in any material respect the consummation of the transactions contemplated by this Agreement or impose any new or additional conditions or otherwise expand, amend or modify any other provision of the Debt Commitment Letter that would materially and adversely affect the ability of Purchaser to fund its obligations when due under this Agreement; or
(ii)terminate, or permit the termination of, the Debt Commitment Letter (other than termination upon entering into a Definitive Agreement), unless such Debt Commitment Letter is replaced with a new commitment that, were it structured as an amendment to an existing Debt Commitment Letter, would satisfy the requirements of the foregoing clause (i). For the avoidance of doubt, nothing herein shall prevent the Purchaser or its Affiliate, as applicable, from replacing or amending the Debt Commitment Letter in order to add lead arrangers, bookrunners, syndication agents or similar entities which had not executed the Debt Commitment Letter as of the date hereof or as required pursuant to the market flex provisions in the fee letters.
(d)In the event that any portion of the Debt Financing becomes unavailable, regardless of the reason therefor, Purchaser or its Affiliate, as applicable will use its reasonable best efforts to: (i) obtain alternative debt financing (in an amount sufficient for the Purchaser to pay the Purchase Price) from the same or other sources that are on terms that are no less favorable, taken as a whole, to Purchaser or its Affiliate, as applicable, and (ii) promptly notify Seller of such unavailability and the reason therefor. For the purposes of this Agreement, the term Debt Commitment Letter shall be deemed to include any commitment letter (or similar agreement) with respect to any alternative debt financing arranged in compliance herewith (and any Debt Commitment Letters remaining in effect at the time in question), as well as all amendments, modifications and supplements permitted under this Agreement.
(e)Purchaser shall provide Seller with prompt written notice of (i) any material breach or default by any party to the Debt Commitment Letter or the Definitive Agreements of which Purchaser or its Affiliate, as applicable, becomes aware or (ii) the receipt of any notice or other communication from any financing source with respect to any breach, default, termination or repudiation by any party to the Debt Commitment Letter or the Definitive Agreements of any provision thereof (but excluding, for the avoidance of doubt, any ordinary course negotiations with respect to the terms of the Debt Financing or Definitive Agreements); provided, that any information disclosed in such notice shall be subject to the confidentiality covenants set forth in Section 6.3. Notwithstanding the foregoing, compliance by Purchaser with this Section 6.22 shall not relieve Purchaser of its obligation to consummate the transactions contemplated by this Agreement whether or not the Debt Financing is available and Purchaser
54
acknowledges that this Agreement and the transactions contemplated hereby are not contingent on Purchaser’s or its Affiliate’s, as applicable, ability to obtain the Debt Financing (or any alternative financing) or any specific term with respect to such financing.
Section 6.23Financing Cooperation.
(a)Prior to the Closing, Seller shall use reasonable best efforts to provide, and to cause its Affiliates to provide, to Purchaser or its Affiliate, as applicable, and at Purchaser’s sole expense, such cooperation reasonably requested by Purchaser in connection with the Debt Financing including:
(i)assisting with the preparation of customary materials for rating agency presentations, bank information memoranda and similar syndication materials; participating in a reasonable number of meetings (including customary meetings among the finance providers, prospective lenders and investors, and senior management and representatives of the Seller and meetings with rating agencies) and providing customary authorization letters to the financing providers authorizing the distribution of information to prospective lenders or investors
(ii)providing all reasonably available financial information of the type and form customarily included in offering documents used for the syndication of credit facilities of the type to be included in the Debt Financing and as may reasonably be requested in connection with the structuring, arrangement and syndication of the Debt Financing; provided, that the Seller shall not provide any pro forma financial information or information about the Seller on an unconsolidated basis;
(iii)reasonably assisting in (A) the preparation and execution and delivery of one or more credit or other agreements governing the Debt Financing, as well as any security documents, intercreditor documents, certificates or other definitive financing documents in connection with the Debt Financing and (B) the facilitation of pledging of collateral and negotiation of payoff letters and lien releases on the Purchased Assets; and
(iv)providing to Purchaser all documentation and other information reasonably requested by Purchaser to comply with applicable “know your customer” and anti-money laundering rules and regulations (including the USA Patriot Act and the Lenders’ corresponding internal policies of general application to all borrowers), to the extent reasonably requested by Purchaser in connection with the Debt Financing at least 10 Business Days prior to Closing;
provided, however, that in no event shall cooperation by Seller be required to the extent it would, or would be reasonably likely to, (A) interfere unreasonably with the business or operations of the Seller, (B) require the Seller to take any action that will conflict with or violate the Seller’s constitutional documents or any applicable Law, (C) require the Seller to enter into or approve any documentation referred to in paragraph (iii) above that takes effect or is effective prior to the Closing or (D) require the Seller to bear any out of pocket cost or expense, pay any fee or provide any indemnity, in each case to the extent effective prior to the Closing.
55
(b)Purchaser or its Affiliate, as applicable, shall, promptly upon request by Seller, reimburse Seller and its Affiliates and each of their respective representatives for all reasonable and documented out of pocket costs and expenses incurred in connection with the cooperation contemplated by this Section 6.23 (including reasonable and documented legal and accounting fees and expenses). Purchaser or its Affiliate, as applicable, shall indemnify and hold harmless Seller and its Affiliates from and against any and all Losses suffered or incurred by it in connection with the arrangement of the Debt Financing (including any information utilized in connection therewith in each case prior to the Closing occurring), except to the extent that such Losses arise out of or in connection with the willful misconduct, bad faith or Fraud by Seller or any of its Affiliates.
Section 6.24Guaranty. Parent Guarantor shall cause Purchaser to timely perform and fulfill its obligations (including payment obligations) under this Agreement and the Ancillary Agreements if, as and when due and shall perform the obligations of Purchaser if Purchaser is unable to, or does not, promptly perform its obligations under this Agreement or any Ancillary Agreement. Parent Guarantor hereby irrevocably and unconditionally guarantees the complete, timely and full discharge by Purchaser of all of its covenants, agreements, obligations and Liabilities under this Agreement and the Ancillary Agreements, including the due and punctual payment of all amounts which are or may become due and payable by Purchaser hereunder or thereunder if, when and as the same shall become due and payable (collectively, the “Obligations”), in accordance with the terms hereof; provided, that in no event shall Parent Guarantor’s aggregate liability hereunder exceed the Purchase Price (the “Guaranty Cap”). Parent Guarantor acknowledges and agrees that, with respect to all Obligations to pay money, such guaranty shall be a guaranty of payment and performance and not of collection and shall not be conditioned or contingent upon the pursuit of any remedies against Purchaser. If Purchaser shall default in the due and punctual performance of any Obligations, including the full and timely payment of any amount due and payable pursuant to any Obligations, Parent Guarantor will forthwith perform or cause to be performed such Obligations and will forthwith make full payment of any amount due with respect thereto at its sole cost and expense, subject to the Guaranty Cap. The guaranty set forth in this Section 6.24 is absolute and unconditional and Parent Guarantor waives any and all defenses available to a guarantor (other than performance in full by Purchaser) that would not be available to Parent Guarantor if Parent Guarantor was the purchasing entity under this Agreement or any Ancillary Agreement instead of Purchaser. Notwithstanding anything to the contrary contained in this Section 6.24 or otherwise, the Parties hereby agree that (a) to the extent Purchaser is relieved of any of its obligations under this Agreement or any Ancillary Agreement, Parent Guarantor shall be similarly relieved of its corresponding obligations under this Section 6.24, (b) Parent Guarantor shall have all rights, remedies, set-offs and defenses to the payment of the Obligation that would be available to Purchaser under this Agreement or any Ancillary Agreement, as well as any defenses in respect of any Fraud of the Seller or its Affiliates and (c) any payment made by or on behalf of Purchaser with respect to the Obligations shall reduce the Obligations of the Parent Guarantor under this Section 6.24 accordingly.
Section 6.25License. Effective as of the Closing, Seller hereby grants to Purchaser an irrevocable and perpetual non-exclusive, worldwide, fully paid-up, freely transferable (solely to Affiliates or Third Parties to which Purchaser is transferring rights to Exploit Subject Products), royalty-free right and license, with the right to grant sublicenses in multiple tiers (solely to
56
Affiliates or Third Parties to which Purchaser is sublicensing rights to sell Subject Products), under all Intellectual Property and Know-How (other than Purchased Intellectual Property and Purchased Know-How) Controlled by Seller or its Affiliates as of the Closing Date (or after the Closing Date to the extent such Intellectual Property and Know-How Controlled by Seller or its Affiliates arises under the Transitional Services Agreement) that is necessary to Exploit Subject Products and for the sole purpose of, and solely to the extent necessary for, Exploiting Subject Products.
Section 6.26Technology Transfer Payments. From and after the Closing Date, Seller shall promptly reimburse Purchaser for any and all payments owed and unpaid by or on behalf of Seller to Patheon Manufacturing Services LLC in connection with technology transfer services related to the Product manufactured prior to the Closing Date within 15 Business Days following Purchaser’s receipt of an invoice therefor, with reasonable supporting documentation.
Section 6.27Patents; Licensing; Royalty Buy-Out.
(a)From and after the Closing Date, Purchaser shall maintain all the Purchased Intellectual Property that are Patents that contain issued and unexpired Valid Claims, and use diligent efforts to prosecute in good faith all Purchased Intellectual Property that are Patents that are pending patent applications.
(b)Unless consented to by the Seller (such consent not to be unreasonably withheld, conditioned or delayed), neither Purchaser nor any of its Affiliates shall [***].
(c)Prior to Closing, the Parties shall discuss and consider a mechanism and valuation method that would provide the Purchaser with a right to buy-out the remaining Royalty Term in the future. If the terms and conditions of such a mechanism are mutually agreed (in each Party’s sole discretion), the Parties would expect to amend and restate this Agreement in order to make corresponding adjustments to the provisions relating to payment of the Royalty and the consent requirement under Section 6.27(b).
Section 6.28SVB Consent; UCC amendments.
(a)Seller (i) shall cause each of the conditions in Section 3 of the SVB Consent to be satisfied on the Closing Date and (ii) shall not, without the prior written consent of Purchaser (not to be unreasonably withheld, conditioned or delayed), permit (x) the termination of the SVB Consent or (y) any material amendment, replacement, supplement or modification to, or any material waiver of any provision under, the SVB Consent if such amendment, modification, waiver or remedy could be reasonably expected to prevent, impede or delay satisfaction of the conditions in Section 3 of the SVB Consent or the effectiveness of the consent granted thereunder, in each case, prior to the Closing Date.
(b)On the Closing Date, Seller shall file UCC amendments, in form reasonably satisfactory to the Purchaser, expressly excluding the Purchased Assets from the description of collateral in the financing statements on record with respect to the Lien granted under the Loan Agreement (as defined in the SVB Consent).
* [***] CERTAIN IDENTIFIED INFORMATION HAS BEEN EXCLUDED FROM THE EXHIBIT BECAUSE IT BOTH (A) IS NOT MATERIAL AND (B) WOULD BE COMPETITIVELY HARMFUL IF PUBLICLY DISCLOSED
57
Section 6.29Replicative Agreements. From and after the Closing Date, Seller shall use commercially reasonable efforts to assist Purchaser in negotiating agreements between Purchaser and each of the counterparties to the Contracts listed on Section 6.29 of the Seller Schedule (each, a “Retained Shared Contract”), such agreements to be on substantially the same terms and conditions as the Retained Shared Contracts. Until the earlier of such time as Purchaser enters into such an agreement with respect to each Retained Shared Contract and one year following the Closing Date, such Retained Shared Contract shall be deemed to be a “Shared Contract” for purposes of Section 2.5(c) of this Agreement (except that such Retained Shared Contract shall not be assigned, transferred or otherwise conveyed by Seller to Purchaser).
ARTICLE VII
Conditions to Closing
Section 7.1Conditions to Each Party’s Obligations. The obligations of Purchaser and the Seller to consummate the Closing are subject to the satisfaction (or waiver in writing by the Party whose obligations to consummate the Closing are subject thereto) at or prior to the Closing of the following conditions:
(a)Governmental Approvals. Any applicable waiting period under the HSR Act shall have expired or been terminated.
(b)No Injunctions or Restraints. No Governmental Authority of competent jurisdiction shall have enacted, issued, promulgated or enforced any applicable Law or preliminary or permanent injunction or order which is in effect and which prohibits, enjoins or otherwise restrains the Acquisition and no Action seeking any such injunction or order shall have been filed with any Governmental Authority, which Action remains pending as of the Closing.
Section 7.2Conditions to Obligation of Purchaser. The obligation of Purchaser to consummate the Closing is subject to the satisfaction (or waiver, to the extent permitted by applicable Law, by Purchaser) at or prior to the Closing of the following conditions:
(a)Representations and Warranties. (i) The Seller Specified Representations shall be true and correct in all material respects at the Closing as though made as of the Closing (except, in each case, to the extent that such representation and warranty speaks only as of a particular date, in which case such representation and warranty shall be true and correct in all material respects as of such particular date) and (ii) the representations and warranties of the Seller set forth in this Agreement (other than the Seller Specified Representations) shall be true and correct (without giving effect to any “materiality” or “Business Material Adverse Effect” qualifiers contained therein) at the Closing as though made as of the Closing (except, in each case, to the extent that such representation and warranty speaks only as of a particular date, in which case such representation and warranty shall be true and correct as of such particular date), except where the failure of any of such representations and warranties of the Seller to be so true and correct would not reasonably be expected to have a Business Material Adverse Effect.
58
(b)Performance of Obligations of the Seller. The Seller shall have performed or complied in all material respects with all obligations and covenants required by this Agreement to be performed or complied with by it by the time of the Closing.
(c)Deliverables by the Seller. The Seller shall have complied in all respects with the provisions of Section 2.8(a).
(d)Approvals. The Seller shall have obtained consents from the Persons set forth on Section 2.8(a)(xiii) of the Seller Schedule.
(e)No Business Material Adverse Effect. From the Execution Date until the Closing Date, no event or events shall have occurred and be continuing which, individually or in the aggregate, constitute or could reasonably be expected to constitute a Business Material Adverse Effect.
Section 7.3Conditions to Obligation of the Seller. The obligation of the Seller to consummate the Closing is subject to the satisfaction (or waiver, to the extent permitted by applicable Law, by the Seller) on or prior to the Closing Date of the following conditions:
(a)Representations and Warranties. (i) The Purchaser Specified Representations and the representations and warranties of Purchaser set forth in Section 4.9 (Financial Ability) and Section 4.6 (Solvency) shall be true and correct in all material respects at the Closing as though made as of the Closing (except, in each case, to the extent that such representation and warranty speaks only as of a particular date, in which case such representation and warranty shall be true and correct in all material respects as of such particular date) and (ii) the representations and warranties of Purchaser (other than those described in the immediately preceding clause (i)) set forth in this Agreement shall be true and correct (without giving effect to any “materiality” or “Purchaser Material Adverse Effect” qualifiers contained therein) at the Closing as though made as of the Closing (except, in each case, to the extent that such representation and warranty speaks only as of a particular date, in which case such representation and warranty shall be true and correct as of such particular date), except where the failure of any of such representations and warranties of Purchaser to be so true and correct would not reasonably be expected to have a Purchaser Material Adverse Effect.
(b)Performance of Obligations of Purchaser. Purchaser shall have performed or complied in all material respects with all obligations and covenants required by this Agreement to be performed or complied with by Purchaser by the time of the Closing.
(c)Deliverables by Purchaser. Purchaser shall have complied in all respects with the provisions of Section 2.8(b).
Section 7.4Frustration of Closing Conditions. Neither Purchaser nor the Seller may rely on the failure of any condition set forth in this Article VII to be satisfied if such failure was caused by such Party’s failure to act in good faith or to comply with its agreements set forth herein.
ARTICLE VIII
Termination
59
Section 8.1Termination. This Agreement may be terminated and the Acquisition and the other transactions contemplated by this Agreement and the Ancillary Agreements abandoned at any time prior to the Closing:
(a)by mutual written consent of the Seller and Purchaser;
(b)by Seller or Purchaser, by written notice to the other Party:
(i)if the Closing does not occur on or prior to the date that is 120 days following the Execution Date (the “Outside Date”); provided, however, that the right to terminate this Agreement under this Section 8.1(b)(i) shall not be available to any Party whose willful breach or failure to perform any of its obligations under this Agreement has been the cause of, or materially contributed to, the failure of the Closing to have occurred on or before the Outside Date.
(ii)if consummation of the transactions contemplated hereby would violate any non-appealable final order, decree or judgment of any Governmental Authority having competent jurisdiction; provided, however, that the right to terminate this Agreement under this Section 8.1(b)(ii) shall not be available to any Party whose willful breach or failure to perform any of its obligations under this Agreement has been the cause of, or materially contributed to, the issuance of such non-appealable final order, decree or judgment.
(c)by Purchaser, by written notice to the Seller, if the Seller shall have breached or failed to perform any of its representations, warranties, covenants or agreements contained in this Agreement, which breach or failure to perform (i) would give rise to the failure of a condition set forth in Sections 7.1 or 7.2 and (ii) cannot be cured by the Seller by the Outside Date, or if capable of being cured, shall not have been cured by the earlier of (1) the 30th day following receipt by the Seller of written notice of such breach or failure to perform from Purchaser stating Purchaser’s intention to terminate this Agreement pursuant to this Section 8.1(c) and the basis for such termination and (2) the Outside Date; provided, however, that Purchaser shall not have the right to terminate this Agreement pursuant to this Section 8.1(c) if Purchaser is then in breach of any representations, warranties, covenants or other agreements hereunder which breach would result in a condition to Closing set forth in Sections 7.1 or 7.3 not being satisfied; or
(d)by the Seller, by written notice to Purchaser, if Purchaser shall have breached or failed to perform any of its representations, warranties, covenants or agreements contained in this Agreement, which breach or failure to perform (i) would give rise to the failure of a condition set forth in Sections 7.1 or 7.3 and (ii) cannot be cured by Purchaser by the Outside Date, or if capable of being cured, shall not have been cured by the earlier of (A) the 30th day following receipt by Purchaser of written notice of such breach or failure to perform from the Seller stating the Seller’s intention to terminate this Agreement pursuant to this Section 8.1(d) and the basis for such termination and (B) the Outside Date; provided, however, that the Seller shall not have the right to terminate this Agreement pursuant to this Section 8.1(d) if the Seller is then in breach of any representations, warranties, covenants or other agreements hereunder which breach would result in a condition to Closing set forth in Sections 7.1 or 7.2 not being satisfied (other than those conditions that (x) by their terms are to be satisfied at the
60
Closing or (y) the failure of which to be satisfied is attributable primarily to a breach by Purchaser of its representations, warranties, covenants or agreements contained in this Agreement).
Section 8.2Effect of Termination.
(a)In the event of termination by the Seller or Purchaser pursuant to Section 7.1, written notice thereof shall forthwith be given to the other Party, specifying the provision hereof pursuant to which such termination is made, and this Agreement shall forthwith become null and void and of no further force and effect (other than the provisions of Section 6.3 (Confidentiality), Section 6.6 (Publicity), Article VIII (Termination), Section 11.3 (Fees and Expenses), Section 11.4 (Notices), Section 11.5 (Interpretation), Section 11.9 (Governing Law) and Section 11.10 (Waiver of Jury Trial), all of which shall survive termination of this Agreement), and there shall be no liability on the part of Purchaser or Seller or their respective Affiliates or representatives, except as liability may exist pursuant to the sections specified in this Section 8.2(a) that survive such termination.
(b)If the transactions contemplated by this Agreement are terminated as provided herein (i) Purchaser promptly shall, and shall cause each of its controlled Affiliates and representatives who were provided with Confidential Information by Purchaser to, return to the Seller or destroy, all Confidential Information and other documents and other material received from the Seller or any of its Affiliates or their respective representatives relating to the transactions contemplated by this Agreement, whether so obtained before or after the execution hereof; (ii) all information received by Purchaser or its Affiliates or representatives with respect to the business of the Seller and its Affiliates shall be treated in accordance with Section 6.3(a), which the Parties agree shall remain in full force and effect notwithstanding the termination of this Agreement and (iii) as soon as practicable following a termination of this Agreement for any reason, but in no event more than 30 days after such termination, Purchaser and the Seller shall, to the extent practicable, withdraw all filings, applications and other submissions relating to the transactions contemplated by this Agreement filed or submitted by or on behalf of such Party to any Governmental Authority or other person.
ARTICLE IX
Indemnification; Survival
Section 9.1Indemnification by the Seller. Subject to the provisions of this Article IX, from and after the Closing, the Seller shall indemnify Purchaser and its Affiliates and each of their respective officers, directors, representatives and employees (collectively, the “Purchaser Indemnitees”), from and against any and all out-of-pocket losses, damages, charges, claims, fines, penalties, interest or expenses, including reasonable third-party legal fees and expenses in connection with any Action (collectively, “Losses”), suffered, incurred or sustained by a Purchaser Indemnitee to the extent arising or resulting from any of the following:
(a)any breach of any representation or warranty of Seller contained in this Agreement; provided, that all “material,” “materiality,” “Business Material Adverse Effect” or similar qualifications limiting the scope of such representation or warranty shall be disregarded
61
for the purposes of this Article IX (solely for the purposes of determining the amount of Loss resulting from any such breach);
(b)any breach or failure by Seller to perform any covenant or agreement of the Seller contained in this Agreement; and
(c)any Excluded Asset or Retained Liability.
Section 9.2Indemnification by Purchaser. Subject to the provisions of this Article IX, from and after the Closing, Purchaser shall indemnify the Seller and its Affiliates and each of their respective officers, directors representatives and employees (the “Seller Indemnitees”) from and against any and all Losses, actually suffered, incurred or sustained by a Seller Indemnitee, to the extent arising or resulting from any of the following:
(a)any breach of any representation or warranty of Purchaser contained in this Agreement;
(b)any breach or failure by Purchaser to perform any covenant of Purchaser contained in this Agreement; and
(c)any Assumed Liability.
Section 9.3Indemnification Procedures.
(a)Third Party Claims. If any Party (the “Indemnified Party”) receives written notice of the commencement of any Action or the assertion of any claim by a third party or the imposition of any penalty or assessment for which indemnity may be sought under Sections 9.1 or 9.2 (a “Third Party Claim”), and such Indemnified Party intends to seek indemnity pursuant to this Article IX, the Indemnified Party shall promptly provide the other Party (the “Indemnifying Party”) with written notice of such Third Party Claim, stating the nature, basis and the amount thereof, to the extent known, along with copies of the relevant documents evidencing such Third Party Claim and the basis for indemnification sought. Failure of the Indemnified Party to give such notice within the time frame specified will not relieve the Indemnifying Party from its indemnification obligations hereunder, except to the extent that the Indemnifying Party is actually prejudiced thereby. The Indemnifying Party will have 30 days from receipt of any such notice of a Third Party Claim to give notice to assume the defense, appeal or settlement proceedings thereof. If notice to the effect set forth in the immediately preceding sentence is given by the Indemnifying Party, the Indemnifying Party will have the right to assume the defense, appeal or settlement proceedings of the Indemnified Party against the Third Party Claim with counsel of its choice; provided, however, that the Indemnifying Party shall not be entitled to assume the defense of such Third Party Claim if (x) the claim seeks injunctive or other equitable relief or (y) the Indemnified Party shall have reasonably concluded that an actual or potential conflict of interest exists between the Indemnifying Party and the Indemnified Party that would make separate representation inadvisable, including, for the avoidance of doubt, any Third Party Action involving a warranty or other claim in connection with (i) Tax Returns or (ii) a commercial relationship between or involving the third party claimant or its Affiliates, on the one hand, and the Indemnified Party or its Affiliates, on the other hand. The Indemnified Party shall diligently conduct such defense, appeal or settlement
62
(including the making of all filings and responses due during such time) during the period prior to the assumption of such defense, appeal or settlement proceeding by the Indemnifying Party, or the expiration of such 30-day notice period. So long as the Indemnifying Party has assumed the defense, appeal or settlement proceedings of the Third Party Claim in accordance herewith, (i) the Indemnified Party may retain separate co-counsel at its sole cost and expense and participate in the defense, appeal or settlement proceedings of the Third Party Claim, (ii) the Indemnified Party shall not file any papers with respect to the Third Party Claim without the prior written consent of the Indemnifying Party and (iii) the Indemnifying Party shall not admit to any wrongdoing by the Indemnified Party. The Indemnifying Party shall have the right to settle any Third Party Claim for which it obtains a full release of the Indemnified Party with respect to such Third Party Claim or to which settlement the Indemnified Party consents in writing (such consent not to be unreasonably withheld, conditioned or delayed). As to any Third Party Claim with respect to which the Indemnifying Party does not elect to assume control of the defense, the Indemnified Party will afford the Indemnifying Party an opportunity to participate in such defense, at its cost and expense, and will consult with the Indemnifying Party prior to settling or otherwise disposing of any of the same. The Parties will act in good faith in responding to, defending against, settling or otherwise dealing with Third Party Claims. The Parties will also cooperate in any such defense, appeal or settlement proceedings, and give each other reasonable access to all information relevant thereto. Whether or not the Indemnifying Party has assumed the defense, appeal or settlement proceedings with respect to a Third Party Claim, such Indemnifying Party will not be obligated to indemnify the Indemnified Party hereunder for any settlement entered into or any judgment that was consented to without the Indemnifying Party’s prior written consent (such consent not to be unreasonably withheld, conditioned or delayed).
(b)Other Claims.
(i)As soon as reasonably practicable, but in no event later than 30 days, after an Indemnified Party becomes aware of any claim (a “Direct Claim”) that does not involve a Third Party Claim that might result in Losses for which such Indemnified Party may be entitled to indemnification under this Article IX, the Indemnified Party shall provide written notice (a “Claim Notice”) to the Indemnifying Party: stating the nature, basis, the amount thereof (to the extent known or estimated, which amount shall not be conclusive of the final amount of such Direct Claim), the method of computation thereof (to the extent known or estimated) and, to the extent practicable, any other material details pertaining thereto, along with copies of the relevant documents evidencing such Direct Claim and the basis for indemnification sought. Failure of the Indemnified Party to give such Claim Notice within the time frame specified will not relieve the Indemnifying Party from its indemnification obligations hereunder, except to the extent that the Indemnifying Party is actually prejudiced thereby.
(ii)Following receipt of a Claim Notice from an Indemnified Party, the Indemnifying Party shall have 60 days to make such investigation of the claim as the Indemnifying Party reasonably deems necessary or desirable. For the purposes of such investigation, the Indemnified Party agrees to make available to the Indemnifying Party or its representatives the information relied upon by the Indemnified Party to substantiate the claim and all other information in the Indemnified Party’s possession or under the Indemnified Party’s control that the Indemnifying Party reasonably requests.
63
(iii)Within such 60-day period, an Indemnifying Party may object to any claim set forth in such Claim Notice by delivering written notice to the Indemnified Party of the Indemnifying Party’s objection (an “Indemnification Objection Notice”). Such Indemnification Objection Notice must describe the grounds for such objection in reasonable detail. If an Indemnification Objection Notice is not delivered by the Indemnifying Party to the Indemnified Party within 60 days after receipt by the Indemnifying Party of the Claim Notice (the “Indemnification Objection Period”), the Indemnified Party shall be free to seek enforcement of its rights to indemnification under this Agreement with respect to such Direct Claim.
(iv)If an Indemnifying Party shall object in writing during the Indemnification Objection Period to any claim or claims by an Indemnified Party made in any Claim Notice, the Indemnified Party shall have 30 days after its receipt of the Indemnification Objection Notice to respond in a written statement to such objection. If after such 30-day period there remains a dispute as to any claims, the Indemnifying Party and the Indemnified Party shall attempt in good faith for 20 days (or any mutually agreed upon extension thereof) thereafter to agree in writing upon the rights of the respective Parties with respect to each of such claims. If no such written agreement can be reached after good faith negotiation, each of the Indemnifying Party and the Indemnified Party may take action to resolve the objection in accordance with Sections 11.9, 11.10 and 11.11.
Section 9.4Limitations on Indemnification. Notwithstanding anything to the contrary contained in this Agreement:
(a)except with respect to claims for Losses arising from Fraud, Seller shall not be liable for any Losses with respect to the matters set forth in Section 9.1(a) (i) unless the amount of Losses with respect to any individual indemnified matter (together with Losses from any substantially similar event, occurrence, condition or set of facts or circumstances) is greater than $[***] (“Per Claim Threshold”) and (ii) until the aggregate amount of all such Losses exceeds $[***] (the “Basket”); provided, however, that Losses attributable to a breach of any Seller Specified Representations shall not be subject to the Per Claim Threshold or the Basket; and provided, further, however, that if such aggregate amount of Losses exceeds the Basket, then the Purchaser Indemnitee shall be entitled to indemnification for the entire amount of all such Losses.
(b)except with respect to claims for Losses arising from Fraud, Purchaser shall not be liable for any Losses with respect to the matters set forth in Section 9.2(a) until the aggregate amount of all such Losses exceeds the Basket; provided, however, that if such aggregate amount of Losses exceeds the Basket, then the Seller Indemnitee shall be entitled to indemnification for the entire amount of all such Losses.
(c)except with respect to claims for Losses arising from Fraud, the Seller’s maximum liabilities to the Purchaser Indemnitees under Section 9.1(a) shall not exceed an aggregate amount equal to $[***] (the “Cap”); provided, however, that for Losses attributable to a breach of any Seller Specified Representations, the Cap shall equal the Purchase Price.
* [***] CERTAIN IDENTIFIED INFORMATION HAS BEEN EXCLUDED FROM THE EXHIBIT BECAUSE IT BOTH (A) IS NOT MATERIAL AND (B) WOULD BE COMPETITIVELY HARMFUL IF PUBLICLY DISCLOSED
64
(d)except with respect to claims for Losses arising from Fraud, the Purchaser’s maximum liabilities to the Seller Indemnitees under Section 9.2(a) shall not exceed an aggregate amount equal to the Cap; provided, however, that for Losses attributable to a breach of any Purchaser Specified Representations, the Cap shall equal the Purchase Price.
(e)without limitation to the other provisions of this Section 9.4, except with respect to claims for Losses arising from Fraud, the maximum liabilities to the Purchaser Indemnitees or the Seller Indemnitees, as the case may be, under this Article IX shall not exceed an aggregate amount equal to the Purchase Price;
(f)no Party shall have any liability for an otherwise indemnifiable Loss that is contingent unless and until such contingent Loss becomes an actual Loss of the Indemnified Party and is due and payable;
(g)no Party shall be liable for any Losses to the extent the Purchaser Indemnitees or the Seller Indemnitees, as applicable, failed to take reasonable steps to mitigate such Losses or otherwise comply with its duty to mitigate in accordance with Laws and Section 9.5(c);
(h)no Party shall be liable for any Loss to the extent arising from (i) a change in Law or a change in accounting or taxation policy or practice made after the Closing, other than a change required to comply with any Law, policy or practice in effect on the Execution Date, or (ii) any Law not in force on the Execution Date or any change in Law which takes effect retroactively or occurs as a result of any increase in the rates of taxation in force on the Execution Date;
(i)no Party shall be liable for any otherwise indemnifiable Loss arising out of any breach of any representation, warranty, covenant or agreement of such Party unless a timely claim therefore is asserted with specificity and in writing by the Indemnified Party in accordance with Section 9.7, failing which such claim shall be waived and extinguished; and
(j)Losses shall include only direct and actual losses, and shall not include special, indirect, incidental, exemplary, punitive or consequential damages of any Party or lost or anticipated profits, revenues or opportunities or business interruption of any other Party, or any damages calculated by reference to a multiplier of revenue, profits, EBITDA or similar methodology, whether or not caused by or resulting from the actions of a Party or the breach of its covenants, agreements, representations or warranties hereunder and whether or not based on or in warranty, contract, tort (including negligence, strict liability or innocent or negligent misrepresentation or misstatement) or otherwise; provided, however, that nothing in this Section 9.4 shall preclude any recovery by an Indemnified Party against an Indemnifying Party for a Third Party Claim otherwise recoverable hereunder.
Section 9.5Calculation of Indemnity Payments.
(a)The amount of any Loss for which indemnification is provided under this Article IX shall be (i) net of any amounts actually recovered by the Indemnified Party under insurance policies with respect to such Loss, and (ii) reduced to take account of any net Tax benefit actually realized by the Indemnified Party arising from the incurrence or payment of any
65
such indemnified amount. In computing the amount of any such Tax benefit, the Indemnified Party shall be deemed to recognize all other items of income, gain, loss, deduction or credit before recognizing any item arising from the receipt of any indemnity payment hereunder or the incurrence or payment of any indemnified amount.
(b)If an Indemnified Party recovers an amount from a third party in respect of Losses that are the subject of indemnification hereunder after all or a portion of such Losses have been paid by an Indemnifying Party pursuant to this Article IX, then the Indemnified Party shall promptly remit to the Indemnifying Party the excess (if any) of (i) (1) the amount paid by the Indemnifying Party in respect of such Losses plus (2) the amount received by the Indemnified Party in respect thereof over (ii) the full amount of the Losses. In the event that an Indemnified Party has any rights against a third party with respect to any Loss that results in a payment by an Indemnifying Party under this Article IX, such Indemnifying Party shall be subrogated to such rights to the extent of such payment. Without limiting the generality of any other provision hereof, each Indemnified Party shall duly execute upon request all instruments reasonably necessary to evidence and perfect the subrogation and subordination rights detailed herein, and otherwise cooperate in the prosecution of such claims.
(c)Each Party shall, and shall cause its respective Affiliates to, take all reasonable steps to mitigate any Loss indemnifiable hereunder upon and after becoming aware of any event that could reasonably be expected to give rise to any Loss. No Party shall be entitled to any payment, adjustment or indemnification more than once with respect to the same matter.
Section 9.6Exclusivity.
(a)Except (i) as expressly provided for in this Agreement or in the Ancillary Agreements in accordance with their terms and (ii) with respect to Losses resulting from Fraud, from and after the Closing, each Party’s sole and exclusive remedy with respect to any and all claims relating to this Agreement, the Business, the Purchased Assets, the Assumed Liabilities, or the transactions contemplated by this Agreement shall be pursuant to the indemnification provisions set forth in this Article IX and the remedies in Section 11.13, as applicable. In furtherance of the foregoing, each Party hereby waives, from and after the Closing, any and all rights, claims and causes of action whether based on warranty, in contract, in tort (including negligence, strict liability or innocent or negligent misrepresentation or misstatement) or otherwise that such Party or any other Purchaser Indemnitee or Seller Indemnitee, as applicable, may have against the other Party, any of its Affiliates or any other Person, arising under or based upon any Law, except pursuant to the indemnification provisions set forth in this Article IX, the remedies in Section 11.13, and except with respect to Losses resulting from Fraud. Notwithstanding anything to the contrary contained in this Agreement, no breach of any representation, warranty, covenant or agreement contained herein shall give rise to any right on the part of one Party, on the one hand, or the other Party, on the other hand, after the consummation of the transactions contemplated by this Agreement, to rescind this Agreement or any of the transactions contemplated hereby.
(b)Purchaser, on behalf of itself and each other Purchaser Indemnitee, acknowledges and agrees that Sellers’s indemnification obligations under Article IX will be paid solely as follows: (A) first, by way of deduction and set off against any (i) Royalty or (ii)
66
Milestone Payment, in each case, owed by Purchaser to the Seller and (B) second, by direct payment by Seller to the applicable Purchaser Indemnitee (other than with respect to claims for indemnification under Section 9.1(a) for breach of representations and warranties of Seller that are not Seller Specified Representations, which shall only be payable pursuant to clause (A)(i)); provided, that nothing herein shall in any way limit the Purchaser’s recourse against any Person in respect of Fraud.
Section 9.7Tax Treatment of Indemnification. For all Tax purposes, Purchaser and the Seller agree to treat any indemnity payment under this Agreement as an adjustment to the Purchase Price unless otherwise required pursuant to a “determination” under Section 1313(a) of the Code (or corresponding provision of state, local or non-U.S. Tax Laws).
Section 9.8Survival. The representations and warranties of Seller contained in this Agreement shall survive the Closing and shall terminate at the close of business *[***] months following the Closing; provided, however, that (i) the Seller Specified Representations shall survive the Closing and shall terminate at the close of business six years following the Closing and (ii) the representations and warranties contained in Section 3.8 (Taxes) shall survive the Closing and shall terminate 90 days after the expiration of the statute of limitations applicable to the matters covered thereby (giving effect to any extension thereof), or, if no such statute of limitations exists, until the 10-year anniversary of the date hereof. The representations and warranties of Purchaser and Parent Guarantor contained in this Agreement shall survive the Closing solely for purposes of Section 9.2(a) and shall terminate at the close of business one year following the Closing, other than the representation and warranty set forth in Section 4.6 (Solvency) and Section 4.9 (Financial Ability), which shall terminate 90 days after the expiration of the statute of limitations applicable to the matters covered thereby (giving effect to any extension thereof) or, if longer, the fourth anniversary of the Closing. None of the covenants or agreements contained in this Agreement or in any Ancillary Agreement shall survive the Closing other than those which by their terms contemplate performance after the Closing Date. No Party shall have any liability or obligation of any nature with respect to any representation, warranty, agreement or covenant after the termination thereof unless a notice of a breach thereof giving rise to a right of indemnity shall have been given to the Party against whom such indemnity may be sought prior to such time. Notwithstanding anything to the contrary hereunder, if notice of an indemnifiable claim is properly delivered in accordance with the terms of this Agreement prior to the expiration of the applicable survival period, the survival period for such claim shall continue until such claim is fully resolved.
ARTICLE X
Tax matters
Section 10.1Tax Covenants.
(a)Purchase Price Allocation. As soon as reasonably practicable following the Execution Date and prior to the Closing Date, Purchaser and the Seller shall use commercially reasonable efforts to agree on an allocation of the Purchase Price and the Assumed Liabilities among the Purchased Assets; provided, however, that upon the written request of one Party to the other Party, the Parties shall continue to use commercially reasonable efforts to agree on such an allocation for 60 days following the Closing Date. The allocation shall be consistent
* [***] CERTAIN IDENTIFIED INFORMATION HAS BEEN EXCLUDED FROM THE EXHIBIT BECAUSE IT BOTH (A) IS NOT MATERIAL AND (B) WOULD BE COMPETITIVELY HARMFUL IF PUBLICLY DISCLOSED
67
with the Estimated Inventory Value, Section 1060 of the Code and the Treasury Regulations issued thereunder, and the methodology described in Section 10.1(a) of the Seller Schedule. If the Parties agree on such an allocation (such agreed allocation, if any, the “Final Allocation”), The Final Allocation shall thereafter be amended to reflect any adjustments to the Purchase Price or the Assumed Liabilities under this Agreement, including Section 2.7(b), Section 2.7(c) and Section 2.7(i), and shall be consistent with Section 1060 of the Code and the Treasury Regulations thereunder (and any provisions of state and local Tax Law, as appropriate) and the methodology described in Section 10.1(a) of the Seller Schedule. Each of Purchaser and the Seller shall cooperate fully, as and to the extent reasonably requested by the other applicable Party, and shall retain and (upon the other applicable Party’s request) furnish or cause to be furnished to the other applicable Party, as promptly as practicable, such information and assistance relating to the Purchased Assets and the Assumed Liabilities as is reasonably necessary for the preparation and filing of any Tax Return. Purchaser and the Seller agree that, for purposes of applicable Tax Law, each of the Seller, Purchaser and their respective Affiliates shall (i) prepare and file their respective Tax Returns that are filed after the Closing Date on a basis consistent with the Final Allocation (as adjusted, if applicable); (ii) to the extent required by applicable Law, file a duly completed IRS Form 8594 (Asset Allocation Statement) consistent with the Final Allocation and amend such IRS Form 8594 if the Final Allocation is adjusted from time to time; (iii) take no position inconsistent with the Final Allocation (as adjusted, if applicable) in any Tax Proceeding unless otherwise required by applicable Law or required as a result of a “determination” within the meaning of Section 1313 of the Code (or similar provision of state, local or non-U.S. Tax Law); (iv) notify the other Party of any notice from any Taxing Authority disputing or reasonably expected to dispute the Final Allocation (as adjusted, if applicable); and (v) use commercially reasonable efforts to defend the Final Allocation (as adjusted, applicable) in any Tax Proceeding in accordance with the procedures for Tax Proceedings set forth in Section 10.3(c), unless otherwise required by applicable Law or required as a result of a “determination” within the meaning of Section 1313 of the Code (or similar provision of state, local or non-U.S. Tax Law). If Purchaser and the Seller cannot agree on a Final Allocation, each of Purchaser and the Seller and their respective Affiliates may separately determine the allocation of the Purchase Price and the Assumed Liabilities among the Purchased Assets in any manner consistent with applicable Tax Law.
(b)Transfer Taxes. Purchaser shall be responsible for paying all Transfer Taxes. The Seller and Purchaser shall cooperate in timely making all filings, returns, reports and forms as may be required in connection with Purchaser’s payment of Transfer Taxes. The Seller and Purchaser, as appropriate, shall execute and deliver all instruments and certificates necessary to enable the other to comply with any filing requirements relating to any such Transfer Taxes.
Section 10.2Cooperation. The Seller and Purchaser shall each reasonably cooperate, and shall cause their respective Affiliates reasonably to cooperate, in preparing and filing all Tax Returns of the Seller or otherwise relating to the Purchased Assets or the Business, including maintaining and making available to each other all records necessary in connection with Taxes and in resolving all disputes and audits with respect to all taxable periods relating to Taxes. The Seller and its Affiliates will need access, from time to time after the Closing Date, to certain accounting and Tax records and information held by Purchaser or its Affiliates to the extent such records and information pertain to the Purchased Assets or the Business prior to the Closing. Therefore, Purchaser shall, and shall cause each of its Affiliates to, (i) use its best efforts to
68
properly retain and maintain such records until such time as the Seller agree that such retention and maintenance is no longer necessary, and (ii) allow the Seller, its Affiliates and their respective agents and representatives to inspect, review and make copies of such records as the Seller may deem necessary or appropriate from time to time pursuant to the procedures set forth in Section 10.3(c); provided, however, that neither Purchaser nor any of its Affiliates shall have the right to inspect, review or make copies of any income Tax Returns of Seller or its Affiliates.
Section 10.3Payment of Taxes and Tax Proceedings.
(a)Straddle Tax Periods. In the case of any Tax with respect to the Business or the Purchased Assets that is assessed with respect to a Straddle Tax Period, the amount of any Taxes based on or measured by sales, use, receipts, or other similar items of the Business for the Pre-Closing Tax Period shall be determined based on an interim closing of the books as of the close of business on the Closing Date, and the amount of any other Taxes of the Business for a Straddle Tax Period which relate to the Pre-Closing Tax Period shall be deemed to be the amount of such Tax for the entire Straddle Tax Period multiplied by a fraction the numerator of which is the number of days in the portion of the Straddle Tax Period ending on the Closing Date and the denominator of which is the total number of days in the Straddle Tax Period. All Straddle Tax Period Taxes not allocated to the Pre-Closing Tax Period pursuant to the foregoing sentence of this Section 10.3(a) shall be allocated to the Post-Closing Tax Period.
(b)Payment of Taxes. Neither Purchaser nor any of its Affiliates shall file an amended Tax Return, or agree to any waiver or extension of the statute of limitations relating to Taxes with respect to the Business or the Purchased Assets for any Pre-Closing Tax Period or a Straddle Tax Period without the prior written consent of the Seller. Purchaser shall (i) prepare and timely file all Tax Returns (other than any income Tax Returns or Washington B&O Tax Returns of the Seller or its Affiliates) with respect to the Business or the Purchased Assets for any Pre-Closing Tax Period or Straddle Tax Period that are required to be filed (taking into account customary extensions in the ordinary course) after the Closing and (ii) pay all Taxes shown as due and payable thereon. All Taxes with respect to the Business or the Purchased Assets that relate to a Straddle Tax Period shall be allocated to the Pre-Closing Tax Period in accordance with Section 10.3(a), and all other Taxes with respect to the Business or the Purchased Assets that relate to a Straddle Tax Period shall be allocated to the Post-Closing Tax Period. If at the time of the applicable Closing the rate of Tax or the assessed valuation for the taxable period in which the applicable Closing occurs has not yet been fixed, Taxes imposed on a periodic basis shall be prorated based on the rate of Tax and assessed valuation established for the immediately preceding taxable period. The Seller shall reimburse Purchaser for any Taxes paid by Purchaser pursuant to this Section 10.3(b) in respect of a Pre-Closing Tax Period or a Straddle Tax Period that is a Retained Liability, and Purchaser shall reimburse Seller for any Taxes paid by Seller or any of its Affiliates pursuant to this Section 10.3(b) in respect of a Post-Closing Tax Period or a Straddle Tax Period that is not a Retained Liability. Purchaser shall promptly pay over to the Seller any refund of Tax received by Purchaser (including by way of a credit against other Tax liabilities) that is an Excluded Asset.
(c)Tax Proceedings. The Seller may choose in its sole discretion (at its expense) to control, or cause its Affiliates to control, all Tax Proceedings relating to a Pre-Closing Tax Period and any Taxes that are Retained Liabilities, and may make all decisions
69
taken in connection with such Tax Proceeding (including selection of counsel and whether to forego any and all administrative appeals, proceedings, hearings and conferences with any Taxing Authority with respect thereto). Each of Purchaser and the Seller shall provide the other with such information and records, and make such of its officers, directors, employees and agents available during normal business hours, as may reasonably be requested by such other party in connection with the preparation of any Tax Return or the conduct of any Tax Proceeding, the Purchased Assets or the Business for any Pre-Closing Tax Period or a Straddle Tax Period. Notwithstanding anything herein to the contrary, the Seller shall not be required to provide Purchaser with a copy of, or otherwise disclose the contents of, any income Tax Return of the Seller or any of its Affiliates.
ARTICLE XI
Miscellaneous
Section 11.1Assignment. No Party may assign any of its rights under this Agreement without the prior consent of the other Party; provided, that (a) Purchaser and Parent Guarantor may each (i) assign any or all of its respective rights and interests hereunder to one or more of its Affiliates and (ii) designate one or more of its Affiliates to perform its obligations hereunder; provided, however, that such assignment does not result in any greater Taxes than Seller or any of its Affiliates would otherwise have incurred (provided, further, that in making any such determination, no deduction under Section 250 of the Code shall be taken into consideration); (b) Purchaser and Parent Guarantor may each assign any or all of its respective rights and interests hereunder solely for collateral security purposes in connection with the Debt Financing to any Lenders; and (c)(i) the Seller may assign any of its rights under this Agreement to one or more of its Affiliates or designate one or more of its Affiliates to perform any of its respective obligations hereunder and (ii) after the Closing, a Party may assign this Agreement (and any of its rights, interests or obligations hereunder) to any purchaser of all or a substantial portion of its assets or any successor by operation of law so long as such purchaser or successor agrees to satisfy such Party’s obligations under this Agreement. In all cases, the assigning party shall remain responsible for the performance of all of its obligations hereunder notwithstanding such assignment. This Agreement will apply to, be binding in all respects upon, and inure to the benefit of, the successors and permitted assigns of the Parties.
Section 11.2No Third-Party Beneficiaries. Except as provided in Sections 9.1 and 9.2, which confer benefits on certain persons who are not party to this Agreement, this Agreement shall not confer any rights or remedies upon any Person other than the Parties and their respective successors and permitted assigns. Notwithstanding the foregoing, the Lenders are intended beneficiaries of Section 11.10 (Waiver of Jury Trial), Section 11.12 (Amendments and Waivers), Section 11.15 (Debt Financing Parties), and this Section 11.2 and shall have the right to rely on and enforce the rights of the Lenders under such Sections.
Section 11.3Fees and Expenses. Except as otherwise expressly set forth in this Agreement, all fees and expenses (including legal and accounting fees and expenses) incurred in connection with this Agreement and the transactions contemplated hereby shall be paid by the Party incurring such fees and expenses.
70
Section 11.4Notices. All notices, requests, consents or other communications required or permitted under this Agreement shall be in writing and shall be deemed to have been duly given or delivered by any Party (a) upon receipt if delivered personally, (b) on the date of transmission when sent by electronic mail before 5:00 p.m. Local time at the recipient’s location on a Business Day (and otherwise on the next Business Day), (c) one Business Day after being sent by internationally recognized overnight delivery service or (d) two Business Days after being mailed by first-class mail, postage prepaid, and in each case, addressed as follows:
if to the Seller:
Omeros Corporation
201 Elliott Avenue West
Seattle, WA 98119
Phone: 206-676-5000
Attention: General Counsel
Email: generalcounsel@omeros.com
with a copy to (which shall not constitute notice):
Covington & Burling LLP
415 Mission Street
San Francisco, CA 94105
Attention:Ingrid Rechtin
Email:irechtin@cov.com
if to Purchaser:
Rayner Surgical Inc.
The Ridley Innovation Centre
10 Dominion Way, Worthing,
West Sussex, BN148AQ, United Kingdom
Phone: +44(019) 032-58900
Attention: Legal Counsel
Email: alanhemmant@rayner.com
with a copy to (which shall not constitute notice):
Freshfields Bruckhaus Deringer US LLP
601 Lexington Avenue
New York, NY 10022
Attention:Adam H. Golden
Email:adam.golden@freshfields.com
or to such other address(es) or Person as any Party may have specified in a notice duly given to the other Party as provided herein.
Section 11.5Interpretation.
71
(a)No reference to or disclosure of any matter or item in this Agreement or in the Seller Schedule or the Purchaser Schedule: (i) shall be construed as an admission or indication that such matter or item is material or that such matter or item is required to be referred to or disclosed in this Agreement, nor shall it be deemed to establish a standard of materiality now or in the future (it being the intent that no Party nor its Affiliates shall be penalized for having disclosed more than may be required by the request); (ii) represents a determination by the Seller or Purchaser, as applicable, or any of their respective Affiliates, that such matter or item did not arise in the ordinary course; (iii) shall imply that such matter or item constitutes or would result in a Business Material Adverse Effect or Purchaser Material Adverse Effect, as applicable, by the criteria set forth in this Agreement; or (iv) shall imply that disclosure of any such matter or item is required by Law or by any Governmental Authority. Without limiting the foregoing, no such reference to or disclosure of a possible breach or violation of any Contract, Law or Judgment shall be construed as an admission or indication that a breach or violation exists or has actually occurred. The Seller and Purchaser agree that the specification of any dollar amount in the representations and warranties or covenants contained in this Agreement or the inclusion of any specific item in the Seller Schedule or Purchaser Schedule is not intended to imply that such amounts or higher or lower amounts, or the items so included or other items, are or are not material, and no Party shall use the fact of the setting of such amounts or the fact of the inclusion of any such item in the Seller Schedule or Purchaser Schedule in any dispute or controversy between the Parties as to whether any obligation, item or matter not described in this Agreement or included in the Seller Schedule or Purchaser Schedule is or is not material for purposes of this Agreement.
(b)All Exhibits and Schedules annexed hereto or referred to herein, the Seller Schedule and the Purchaser Schedule, are hereby incorporated in and made a part of this Agreement as if set forth in full herein. Any capitalized terms used in the Seller Schedule and the Purchaser Schedule, as applicable, or in any Exhibit or Schedule annexed hereto but not otherwise defined therein, shall have the meaning as defined in this Agreement.
(c)References to defined terms in the singular shall include the plural and references to defined terms in the plural shall include the singular. “Extent” in the phrase “to the extent” means the degree to which a subject or other thing extends, and such phrase does not mean simply “if.” “Including” (and, with correlative meaning, “include”) means including, without limiting the generality of any description preceding or succeeding such term and the rule of ejusdem generis will not be applicable to limit a general statement preceded, followed by or referable to an enumeration of specific matters, to matters similar to those specifically mentioned.
(d)The descriptive headings of the several Articles and Sections of this Agreement, the Table of Contents to this Agreement, the Seller Schedule and the Purchaser Schedule are inserted for convenience only, do not constitute a part of this Agreement and shall not affect in any way the meaning or interpretation of this Agreement. All references herein to “Articles,” “Sections,” “Exhibits” or “Schedules” shall be deemed to be references to Articles or Sections of this Agreement or Exhibits or Schedules hereto unless otherwise indicated. The terms “hereof,” “herein,” “hereby” and derivative or similar words refer to this entire Agreement. Unless otherwise specified or where the context otherwise requires, (i) wherever used, the word “or” is used in the inclusive sense (and/or), (ii) references to a Person are also to its permitted
72
successors and assigns, (iii) references to a Law include any amendment or modification to such Law and any rules or regulations issued thereunder, in each case, as in effect at the relevant time of reference thereto, and (iv) references to monetary amounts are denominated in United States Dollars. All accounting terms used herein and not expressly defined herein shall have the meanings given to them under US GAAP, unless otherwise specified. Any reference in this Agreement to “made available” means a document or other item of information that was provided to Purchaser and its representatives in the Data Room at least one hour prior to release of signature pages of Purchaser and Seller hereto.
(e)The Seller and Purchaser agree that they have been represented by counsel during the negotiation, drafting, preparation and execution of this Agreement and, therefore, waive the application of any Law or rule of construction providing that ambiguities in a contract or other document will be construed against the Party drafting such contract or document.
Section 11.6Counterparts. This Agreement may be executed in counterparts, each of which shall be deemed an original but all of which together shall constitute one and the same instrument. This Agreement and any amendments hereto, to the extent signed and delivered by means of digital imaging and electronic mail or a facsimile machine, shall be treated in all manner and respects as an original contract and shall be considered to have the same binding legal effects as if it were the original signed version thereof delivered in person.
Section 11.7Entire Agreement.
(a)This Agreement, including the Exhibits, Schedules, Seller Schedule, and Purchaser Schedule and the Ancillary Agreements constitute the entire understanding and agreement between the Parties and supersede any prior understandings, agreements or representations by or among the Parties, written or oral, that may have related in any way to the subject matter hereof. In the event of any conflict between the provisions of this Agreement (including the Seller Schedule, Purchaser Schedule, and Exhibits), on the one hand, and the provisions of the Ancillary Agreements (including the schedules and exhibits thereto), on the other hand, the provisions of this Agreement shall control, except where the respective Ancillary Agreement specifically provides that any specified language therein supersedes any specified language in this Agreement.
(b)The Parties agree to define their rights, liabilities and obligations with respect to such understanding and the transactions contemplated hereby exclusively in contract pursuant to the express terms and provisions of this Agreement, and the Parties expressly disclaim that they are owed any duties or are entitled to any remedies not expressly set forth in this Agreement.
Section 11.8Severability. Any term or provision of this Agreement that is invalid or unenforceable in any situation in any jurisdiction shall not affect the validity or enforceability of the remaining terms and provisions hereof or the validity or enforceability of the offending term or provision in any other situation or in any other jurisdiction. Any provision of this Agreement held invalid or unenforceable only in part or degree will remain in full force and effect to the extent not held invalid or unenforceable.
73
Section 11.9Governing Law. This Agreement shall be governed by and construed in accordance with the Laws of the State of New York, excluding any conflicts or choice of Law rule or principle that might otherwise refer construction or interpretation of this Agreement to the substantive Law of another jurisdiction.
Section 11.10Waiver of Jury Trial. EACH PARTY HEREBY WAIVES, TO THE FULLEST EXTENT PERMITTED BY APPLICABLE LAW, ANY RIGHT IT MAY HAVE TO A TRIAL BY JURY IN RESPECT TO ANY LITIGATION DIRECTLY OR INDIRECTLY ARISING OUT OF, UNDER OR IN CONNECTION WITH THIS AGREEMENT OR ANY TRANSACTION CONTEMPLATED HEREBY. EACH PARTY (A) CERTIFIES THAT NO REPRESENTATIVE, AGENT OR ATTORNEY OF THE OTHER PARTY HAS REPRESENTED, EXPRESSLY OR OTHERWISE, THAT SUCH OTHER PARTY WOULD NOT, IN THE EVENT OF LITIGATION, SEEK TO ENFORCE THE FOREGOING WAIVER AND (B) ACKNOWLEDGES THAT IT AND THE OTHER PARTY HAVE BEEN INDUCED TO ENTER INTO THIS AGREEMENT, BY, AMONG OTHER THINGS, THE MUTUAL WAIVERS AND CERTIFICATIONS IN THIS SECTION 11.10.
Section 11.11Venue. Notwithstanding anything to the contrary in this Agreement, each of the Seller and Purchaser, on behalf of themselves, their respective Subsidiaries and each of their respective controlled Affiliates hereby: (a) agrees that any Action, whether in law or in equity, whether in contract or in tort or otherwise arising out of or relating to, this Agreement or any of the Ancillary Agreements shall be subject to the exclusive jurisdiction of any federal or state court in the Borough of Manhattan, New York, New York and any appellate court thereof and irrevocably submits itself and its property with respect to any such Action to the exclusive jurisdiction of such court, (b) agrees not to bring or support or permit any of its controlled Affiliates to bring or support any Action of any kind or description, whether in law or in equity, whether in contract or in tort or otherwise arising out of or relating to this Agreement or any of the Ancillary Agreements in any forum other than the United States District Court for the Southern District of New York or the Supreme Court of the State of New York sitting in the Borough of Manhattan, and any appellate court from any thereof, and (c) irrevocably waives, to the fullest extent that it may effectively do so, the defense of an inconvenient forum to the maintenance of such Action in any such court.
Section 11.12Amendments and Waivers. This Agreement may not be amended except by an instrument in writing signed on behalf of each of the Parties. Any waiver of any of the terms or conditions of this Agreement must be in writing and must be duly executed by or on behalf of the Party to be charged with such waiver. Except as expressly set forth in this Agreement, the failure of a Party to exercise any of its rights hereunder or to insist upon strict adherence to any term or condition hereof on any one occasion shall not be construed as a waiver or deprive that Party of the right thereafter to insist upon strict adherence to the terms and conditions of this Agreement at a later date. Further, no waiver of any of the terms and conditions of this Agreement shall be deemed to or shall constitute a waiver of any other term of condition hereof (whether or not similar). Notwithstanding anything to the contrary contained in this Agreement, each of Section 11.10 (Waiver of Jury Trial), Section 11.15 (Debt Financing Parties), Section 11.2 (No Third Party Beneficiaries) and this Section 11.12 solely as such Sections relate to the rights or obligations of the Lenders may not be amended, supplemented,
74
waived or otherwise modified in a manner that is materially adverse to a Lender without the prior written consent of such Lender.
Section 11.13Remedies; Specific Performance.
(a)The Parties agree that irreparable damage for which monetary damages, even if available, would not be an adequate remedy, may occur in the event that any of the Parties do not perform their obligations under the provisions of this Agreement or the Ancillary Agreements (including failing to take such actions as are required of them hereunder to consummate this Agreement or the Ancillary Agreements) in accordance with its specified terms or otherwise breach such provisions. Subject to Section 11.13(b), the Parties acknowledge and agree that (i) the Parties shall be entitled to seek an injunction or injunctions, specific performance or other equitable relief to prevent breaches of this Agreement or the Ancillary Agreements and to enforce specifically the terms and provisions hereof or thereof in any court of competent jurisdiction without proof of damages or otherwise, this being in addition to any other remedy to which they are entitled under this Agreement and (ii) the right of specific enforcement is an integral part of the transactions contemplated by this Agreement and without that right, none of the Seller or the Selling Affiliates nor Purchaser would have entered into this Agreement. If any Party brings any Action to enforce specifically the performance of the terms and provisions of this Agreement by the other Party, the Outside Date shall automatically be extended for so long as the Party bringing such Action is actively seeking a court order for an injunction or injunctions or to specifically enforce the terms and provisions of this Agreement. The Parties agree not to assert that a remedy of specific enforcement is unenforceable, invalid, contrary to Law or inequitable for any reason, and not to assert that a remedy of monetary damages would provide an adequate remedy or that the Parties otherwise have an adequate remedy at law. The Parties acknowledge and agree that any Party seeking an injunction or injunctions to prevent breaches of this Agreement and to enforce specifically the terms and provisions of this Agreement in accordance with this Section 11.13 shall not be required to provide any bond or other security in connection with any such order or injunction.
(b)If a court rules that a Party breached its obligations under this Agreement in connection with its failure to effect the Closing in accordance with Section 2.6, but declines to effect the Closing in accordance with Section 2.6, on the terms and subject to the conditions in this Agreement, pursuant to a claim for specific performance brought against such Party pursuant to this Section 11.13, then, in addition to the right of the such Party to terminate this Agreement pursuant to Section 8.1, such Party shall have the right to be paid all costs and expenses (including attorneys’ fees) of such Party in connection with all actions to seek specific performance of the other Party’s obligations pursuant to this Agreement and all actions to collect such fee or expenses. For the avoidance of doubt, in no event shall the exercise of a Party’s right to seek specific performance pursuant to this Section 11.13 reduce, restrict or otherwise limit such Party’s right to terminate this Agreement pursuant to Section 8.1.
(c)Notwithstanding anything contained in this Agreement to the contrary, it is acknowledged and agreed that the Seller shall be entitled to specifically enforce Purchaser’s obligation to effect the Closing on the terms and conditions set forth in this Agreement, but only if (i) all of the conditions set forth in Article VII have been satisfied or waived by the Party entitled to waive (other than those conditions that, by their terms, cannot be satisfied until
75
Closing), (ii) Seller has irrevocably confirmed in a written notice to Purchaser that if specific performance is granted and the Debt Financing is funded and Purchaser complies with its obligations to effect the Closing pursuant to the terms of this Agreement, then the Closing will occur, (iii) the amounts committed to be funded under the Debt Commitment Letter have been funded, (iv) the Inside Date has passed and (v) Purchaser fails to consummate the Closing on or prior to the date required for Closing under this Agreement. Notwithstanding anything in this Agreement to the contrary: (x) no person other than the Seller shall be entitled to seek specific performance of this Agreement against Purchaser; and (y) none of the Seller, its Affiliates, their respective stakeholders or any other Person shall be entitled to specific performance to cause Purchaser to enforce the terms of the Debt Commitment Letter.
Section 11.14Further Assurances. In case at any time after the Closing any further action is necessary to carry out the purposes of this Agreement, each of the Parties shall take such further action (including the execution and delivery of such further instruments and documents) as the other Party may reasonably request, at the sole cost and expense of the requesting Party, except that the performing Party shall bear the cost to the extent such action is an express obligation of the performing Party under this Agreement or any document or agreement contemplated hereby.
Section 11.15Debt Financing Parties. Notwithstanding anything to the contrary in this Agreement, each of the Seller and Purchaser, on behalf of themselves, their respective Subsidiaries and each of their respective controlled Affiliates hereby: (a) agrees that, except as specifically set forth in the documents relating to the Debt Financing, any Action, whether in law or in equity, whether in contract or in tort or otherwise, involving the Debt Financing Parties, arising out of or relating to, this Agreement, the Debt Financing or any of the agreements (including the Debt Commitment Letter) entered into in connection with the Debt Financing or any of the Transactions or the performance of any services thereunder shall be subject to the exclusive jurisdiction of any federal or state court in the Borough of Manhattan, New York, New York and any appellate court thereof and irrevocably submits itself and its property with respect to any such Action to the exclusive jurisdiction of such court, (b) agrees that, except as specifically set forth in the documents relating to the Debt Financing, any such Action shall be governed by the laws of the State of New York (without giving effect to any conflicts of law principles that would result in the application of the laws of another state), except as otherwise provided in the Debt Commitment Letter or other applicable definitive document relating to the Debt Financing, (c) except as specifically set forth in the documents relating to the Debt Financing, agrees not to bring or support or permit any of its controlled Affiliates to bring or support any Action of any kind or description, whether in law or in equity, whether in contract or in tort or otherwise, against any Debt Financing Party in any way arising out of or relating to this Agreement, the Debt Financing, the Debt Commitment Letter or any of the transactions contemplated hereby or the performance of any services thereunder in any forum other than the United States District Court for the Southern District of New York or the Supreme Court of the State of New York sitting in the Borough of Manhattan, and any appellate court from any thereof, (d) irrevocably waives, to the fullest extent that it may effectively do so, the defense of an inconvenient forum to the maintenance of such Action in any such court, (e) agrees that service of process upon such Party, its Subsidiaries or its controlled Affiliates in any such Action shall be effective if notice is given in accordance with this Agreement, (f) knowingly, intentionally and voluntarily waives to the fullest extent permitted by applicable law trial by jury
76
in any Action brought against the Debt Financing Parties in any way arising out of or relating to this Agreement, the Debt Financing, the Debt Commitment Letter or any of the transactions contemplated hereby or the performance of any services thereunder, (g) agrees that none of the Debt Financing Parties will have any liability to the Seller or any of its Subsidiaries or any of their respective Affiliates or representatives (in each case, other than Purchaser or its respective Subsidiaries) relating to or arising out of this Agreement, the Debt Financing, the Debt Commitment Letter or any of the transactions contemplated hereby or the performance of any services thereunder, whether in law or in equity, whether in contract or in tort or otherwise and the Seller (on behalf of itself and its Subsidiaries and Affiliates) agrees not to commence any Action or proceeding against any Debt Financing Party with respect to the foregoing (and in furtherance and not in limitation of the foregoing, the parties acknowledge and agreed that no Debt Financing Party shall be subject to any special, consequential, punitive or indirect damages or damages of a tortious nature) and (h) agrees that the Debt Financing Parties are express third party beneficiaries of, and may enforce, any of the provisions of this Section 11.15, and that such provisions (or any of the defined terms used herein or any other provision of this Agreement to the extent a modification, waiver or termination of such defined term or provision would modify the substance of this Section 11.15) shall not be amended in any way adverse to the Debt Financing Parties without the prior written consent of the Lenders (and any such amendment, waiver or other modification without such prior written consent shall be null and void). For purposes of this Agreement, “Debt Financing Parties” shall mean the Lenders, together with their respective Affiliates and their and their respective Affiliates’ officers, directors, employees, partners, controlling persons, advisors, attorneys, agents and representatives and their respective successors and assigns, in their capacities as such; provided that neither Purchaser nor any Affiliate of Purchaser shall be a Debt Financing Party.
77
IN WITNESS WHEREOF, the parties hereto have duly executed this Agreement as of the date first written above.
| RAYNER SURGICAL INC. | ||
| | | |
| | by: | /s/ Darren Millington |
| | | Name: Darren Michael Millington |
| | | Title: Director |
| | OMEROS CORPORATION | |||
| | | |||
| | | by: | /s/ Gregory Demopulos | |
| | | | Name: Gregory A. Demopulos, M.D. | |
| | | | Title: Chairman and Chief Executive Officer | |
| Solely for the purposes of Article V and Section 6.24: | ||
| | ||
| RAYNER SURGICAL GROUP LIMITED | ||
| | ||
| | by: | /s/ Darren Millington |
| | | Name: Darren Michael Millington |
| | | Title: Chief Financial Officer |
Exhibit 10.44
CONSENT AND SECOND AMENDMENT
TO
LOAN AND SECURITY AGREEMENT
This Consent and Second Amendment to Loan and Security Agreement (this “Amendment”) is entered into this 1st day of December, 2021, by and between SILICON VALLEY BANK (“Bank”) and OMEROS CORPORATION, a Washington corporation (“Borrower”) whose address is 201 Elliott Avenue West, Seattle, Washington 98119.
RECITALS
A.Bank and Borrower have entered into that certain Loan and Security Agreement dated as of August 2, 2019, as amended by that certain First Amendment to Loan and Security Agreement by and between Bank and Borrower dated as of August 7, 2020 (as the same may from time to time be further amended, modified, supplemented or restated, the “Loan Agreement”).
B.Bank has extended credit to Borrower for the purposes permitted in the Loan Agreement.
C.Borrower has notified Bank that Borrower has entered into or will enter into that certain Asset Purchase Agreement among Borrower, Rayner Surgical Inc., a Delaware corporation (the “Buyer”) and, solely for the purposes of Article V and Section 6.24 of the Purchase Agreement, Rayner Surgical Group Limited, a company limited by shares incorporated under the laws of England, in substantially the form attached hereto as Schedule 2 (the “Purchase Agreement”) pursuant to which, Borrower will sell, transfer and assign the Purchased Assets (as such term is defined in the Purchase Agreement) to the Buyer (the “Asset Sale”).
D.Borrower has requested that Bank (i) consent to the Asset Sale and (ii) amend the Loan Agreement to make certain revisions to the Loan Agreement as more fully set forth herein.
E.Bank has agreed to so amend certain provisions of the Loan Agreement and consent to the Asset Sale, but only to the extent, in accordance with the terms, subject to the conditions and in reliance upon the representations and warranties set forth below.
AGREEMENT
NOW, THEREFORE, in consideration of the foregoing recitals and other good and valuable consideration, the receipt and adequacy of which is hereby acknowledged, and intending to be legally bound, the parties hereto agree as follows:
1.Definitions. Capitalized terms used but not defined in this Amendment shall have the meanings given to them in the Loan Agreement.
2.Release of Liens. Subject to the satisfaction of the conditions precedent set forth in Section 3 below on or before the Outside Date (as such term is defined in the Purchase Agreement), as confirmed by Bank in writing (email being acceptable), all liens on the Purchased Assets (as such term is defined in the Purchase Agreement) shall be automatically released, and Borrower or Buyer shall be authorized to file a UCC-3 partial release statement to evidence the
release of Bank’s security interests and liens it may have in the Purchased Assets (as such term is defined in the Purchase Agreement) at Borrower’s sole cost and expense. Nothing contained herein is intended to be, and shall not be construed as, a release or discharge of any liens or security interests on any Collateral other than the Purchased Assets (as such term is defined in the Purchase Agreement).
3.Consent. Bank hereby consents to the Asset Sale and agrees that the Asset Sale shall not, in and of itself, constitute an Event of Default under Section 7.1 of the Loan Agreement (relative to dispositions), provided that such consent is subject to the following conditions being fulfilled, each to the satisfaction of Bank (a) Borrower shall be a surviving legal entity after the consummation of the Asset Sale, (b) the consideration paid by Buyer in connection with the Asset Sale shall consist of an upfront payment in cash by Buyer to Borrower in an amount of One Hundred Twenty-Five Million Dollars ($125,000,000.00) subject to the computations set forth in the Purchase Agreement, (c) Borrower shall not assume nor incur any Indebtedness or Liens in connection with the Asset Sale, (d) the Asset Sale shall occur within one hundred twenty (120) days of the date of this Amendment and Borrower shall provide Bank with a fully-executed copy of the Purchase Agreement contemporaneously with the execution thereof and (e) no Event of Default shall occur or continue, both before and immediately after giving effect to the Asset Sale. The consent provided for herein is a one-time consent relating only to the Asset Sale, and shall not be deemed to constitute an agreement by Bank to any future consent or waiver of the terms and conditions of the Loan Agreement.
4.Amendments to Loan Agreement.
4.1Section 3.4 (Procedure for Borrowing). The penultimate sentence in Section 3.4 is amended in its entirety and replaced with the following:
“In connection with any such notification, Borrower must promptly deliver to Bank by electronic mail or through Bank’s online banking program such reports and information, including without limitation, a Royalty Payment Report, a Borrowing Base Report, sales journals, cash receipts journals, accounts receivable aging reports, as Bank may request in its reasonable discretion.”
4.2Section 5.3 (Accounts Receivable; Royalty Payments). Section 5.3 is amended in its entirety and replaced with the following:
“5.3Accounts Receivable; Royalty Payments.
(a)(i) For each Account with respect to which Advances are requested, on the date each Advance is requested and made, such Account shall be an Eligible Account and (ii) for each royalty payment with respect to which Advances are requested, on the date each Advance is requested and made, such royalty payment shall be an Eligible Monthly Royalty Payment.
(b)All statements made and all unpaid balances appearing in all invoices, instruments and other documents evidencing the Eligible Accounts are and shall be true and correct and all such invoices, instruments and other documents, and all of Borrower’s Books are genuine and in all respects what they
purport to be. All sales and other transactions underlying or giving rise to each Eligible Account shall comply in all material respects with all applicable laws and governmental rules and regulations. Borrower has no knowledge of any actual or imminent Insolvency Proceeding of any Account Debtor whose accounts are Eligible Accounts in any Borrowing Base Report. To the best of Borrower’s knowledge, all signatures and endorsements on all documents, instruments, and agreements relating to all Eligible Accounts are genuine, and all such documents, instruments and agreements are legally enforceable in accordance with their terms.
(c)All statements made and all unpaid balances appearing in all invoices, instruments and other documents evidencing the Eligible Monthly Royalty Payments are and shall be true and correct and all such invoices, instruments and other documents, and all of Borrower’s Books are genuine and in all respects what they purport to be. All sales and other transactions underlying or giving rise to each Eligible Monthly Royalty Payment shall comply in all material respects with all applicable laws and governmental rules and regulations. Borrower has no knowledge of any actual or imminent Insolvency Proceeding of any Account Debtor whose accounts are Eligible Monthly Royalty Payments in any Royalty Payment Report. To the best of Borrower’s knowledge, all signatures and endorsements on all documents, instruments, and agreements relating to all Eligible Monthly Royalty Payments are genuine, and all such documents, instruments and agreements are legally enforceable in accordance with their terms. Borrower is the owner of and has the legal right to sell, transfer, assign and encumber each customer Account, and, there are no defenses, offsets, counterclaims or agreements for which the Account Debtor may claim any deduction or discount.”
4.3Section 6.2 (Financial Statements, Reports, Certificates). Section 6.2 is amended by (i) deleting “and” appearing at the end of subsection (j), (ii) deleting “.” at the end of subsection (k) and inserting “; and” in lieu thereof, and (iii) inserting the following new subsection (l) thereof:
“(l)(i) (A) at all times when a Streamline Period is not in effect or (B) when an Advance is requested and at all times when an Advance is outstanding, within seven (7) days after the end of each month and (ii) (A) at all times when a Streamline Period is not in effect or (B) when an Advance is not outstanding, within forty-five (45) days of the end of each quarter, a royalty payment statement (and any schedules related thereto and including any other information requested by Bank with respect to Borrower’s Eligible Monthly Royalty Payments) including, without limitation, details of Borrower’s Eligible Monthly Royalty Payments including, without limitation, total royalty payments due to Borrower from Rayner, or an affiliate thereof, and copies of written reports delivered by Rayner, or an affiliate thereof, to Borrower specifying royalty calculations and the amount of net revenue from sales of Omidria (including any combination, new formulation, dosages, extensions, next gen and the like) (the “Royalty Payment Report”);”
4.4Section 6.6 (Access to Collateral; Books and Records). Section 6.6 is amended by inserting the following provision to appear as the last sentence thereof:
“Borrower hereby acknowledges that the Post-Sale Audit will be conducted prior to the earlier to occur of (a) the first (1st) Advance after the Second Amendment Effective Date and (b) 180 days following consummation of the Asset Sale.”
4.5Section 7 (Negative Covenants). The Loan Agreement is amended by inserting the following new provision to appear as Section 7.11 thereof:
“7.11Royalty Payment Agreement. (i) Materially alter the royalty payment formulas and calculations set forth in the Purchase Agreement or (ii) amend or modify the terms set forth in that certain agreement by and between Borrower and Rayner, or an affiliate thereof, governing the royalty payments made by Rayner to Borrower.”
4.6Section 13 (Definitions). The definition of “Eligible Accounts” is amended by (i) deleting “and” appearing at the end of subsection (aa), (ii) deleting the “.” at the end of subsection (bb) and inserting “; and” in lieu thereof, and (iii) inserting the following new subsection (cc):
“(cc)Accounts owing from Eligible Monthly Royalty Payments.”
4.7Section 13 (Definitions). The following terms and their respective definitions set forth in Section 13.1 of the Loan Agreement are deleted in their entirety and replaced with the following:
““Borrowing Base” is (a) is eighty-five percent (85.0%) of Eligible Accounts as determined by Bank from Borrower’s most recent Borrowing Base Report (and as may subsequently be updated by Bank based upon information received by Bank including, without limitation, Accounts that are paid and/or billed following the date of the Borrowing Base Report) plus (b) the product of (i) Borrower’s Eligible Monthly Royalty Payments for the most recent month, as determined by Bank from Borrower’s most recent Royalty Payment Report and financial reporting multiplied by (ii) eighty-five percent (85.0%); provided, however, that Bank has the right, after consultation with Borrower, to decrease the foregoing percentages in its good faith business judgment to mitigate the impact of events, conditions, contingencies, or risks which may adversely affect the Collateral or its value.”
4.8Section 13.1 (Definitions). The following new terms and their respective definitions are inserted to appear alphabetically in Section 13.1 of the Loan Agreement:
““Asset Sale” is defined in the Second Amendment.”
““Eligible Monthly Royalty Payments” is the difference of (a) Borrower’s total royalty payments that (i) are pursuant to binding, written agreements which arise in the ordinary course of Borrower’s business, (ii) meets all of Borrower’s representations and warranties described in Section 5.3, (iii) are payable on a monthly basis within 45 days of the invoice date, and (iv) are or may be due and
owing from Rayner or an affiliate thereof minus (b) any discounts, claims, credits, reserves, offsets, adjustments, indemnifications, or other setoffs; provided that Bank reserves the right at any time and from time to time to exclude and/or remove any Account, or portion thereof, from the definition of Eligible Monthly Royalty Payments, in its sole discretion.”
““Post-Sale Audit” is, upon consummation of the Asset Sale, Bank’s inspection of Borrower’s Accounts, the Collateral, and Borrower’s Books, with results satisfactory to Bank in its sole and absolute discretion.”
““Purchase Agreement” is defined in the Second Amendment.”
““Rayner” means, Rayner Surgical Inc., a Delaware corporation.”
““Royalty Payment Report” is defined in Section 6.2(l).”
““Second Amendment” means that certain Consent and Second Amendment to Loan and Security Agreement between Bank and Borrower dated as of December 1, 2021.”
““Second Amendment Effective Date” is December 1, 2021.”
4.9Exhibit B (Compliance Certificate). The Compliance Certificate appearing as Exhibit B to the Loan Agreement is deleted in its entirety and replaced with the Compliance Certificate attached as Schedule 1 attached hereto.
5.Limitation of Amendments.
5.1The amendments set forth in Section 3, above, are effective for the purposes set forth herein and shall be limited precisely as written and shall not be deemed to (a) be a consent to any amendment, waiver or modification of any other term or condition of any Loan Document, or (b) otherwise prejudice any right or remedy which Bank may now have or may have in the future under or in connection with any Loan Document.
5.2This Amendment shall be construed in connection with and as part of the Loan Documents and all terms, conditions, representations, warranties, covenants and agreements set forth in the Loan Documents, except as herein amended, are hereby ratified and confirmed and shall remain in full force and effect.
6.Representations and Warranties. To induce Bank to enter into this Amendment, Borrower hereby represents and warrants to Bank as follows:
6.1Immediately after giving effect to this Amendment (a) the representations and warranties contained in the Loan Documents are true, accurate and complete in all material respects as of the date hereof (except to the extent such representations and warranties relate to an earlier date, in which case they are true and correct as of such date), and (b) no Event of Default has occurred and is continuing;
6.2Borrower has the power and authority to execute and deliver this Amendment and to perform its obligations under the Loan Agreement, as amended by this Amendment;
6.3The organizational documents of Borrower delivered to Bank on the Effective Date remain true, accurate and complete and have not been amended, supplemented or restated and are and continue to be in full force and effect;
6.4The execution and delivery by Borrower of this Amendment and the performance by Borrower of its obligations under the Loan Agreement, as amended by this Amendment, have been duly authorized;
6.5The execution and delivery by Borrower of this Amendment and the performance by Borrower of its obligations under the Loan Agreement, as amended by this Amendment, do not and will not contravene (a) any law or regulation binding on or affecting Borrower, (b) any contractual restriction with a Person binding on Borrower, (c) any order, judgment or decree of any court or other governmental or public body or authority, or subdivision thereof, binding on Borrower, or (d) the organizational documents of Borrower;
6.6The execution and delivery by Borrower of this Amendment and the performance by Borrower of its obligations under the Loan Agreement, as amended by this Amendment, do not require any order, consent, approval, license, authorization or validation of, or filing, recording or registration with, or exemption by any governmental or public body or authority, or subdivision thereof, binding on Borrower, except as already has been obtained or made; and
6.7This Amendment has been duly executed and delivered by Borrower and is the binding obligation of Borrower, enforceable against Borrower in accordance with its terms, except as such enforceability may be limited by bankruptcy, insolvency, reorganization, liquidation, moratorium or other similar laws of general application and equitable principles relating to or affecting creditors’ rights.
7.Ratification of Perfection Certificate. Borrower hereby ratifies, confirms and reaffirms, all and singular, the terms and disclosures contained in a certain Perfection Certificate of Borrower dated as of August 2, 2019 and acknowledges, confirms and agrees the disclosures and information Borrower provided to Bank in said Perfection Certificate have not changed, as of the date hereof.
8.Release by Borrower:
A.FOR GOOD AND VALUABLE CONSIDERATION, Borrower hereby forever relieves, releases, and discharges Bank and its present or former employees, officers, directors, agents, representatives, attorneys (collectively, the “Releasees”), and each of them, from any and all claims, debts, liabilities, demands, obligations, promises, acts, agreements, costs and expenses, actions and causes of action, of every type, kind, nature, description or character whatsoever, whether known or unknown, suspected or unsuspected, absolute or contingent, that Borrower may have against the Releasees which arise out of or in any manner whatsoever connected with or related to facts, circumstances,
issues, controversies or claims existing or arising from the beginning of time through and including the date of execution of this Amendment (collectively “Released Claims”). Without limiting the foregoing, the Released Claims shall include any and all liabilities or claims arising out of or in any manner whatsoever connected with or related to the Loan Documents, the recitals hereto, any instruments, agreements or documents executed in connection with any of the foregoing or the origination, negotiation, administration, servicing and/or enforcement of any of the foregoing.
B.In furtherance of this release, Borrower expressly acknowledges and waives any and all rights under Section 1542 of the California Civil Code, which provides as follows:
“A general release does not extend to claims that the creditor or releasing party does not know or suspect to exist in his or her favor at the time of executing the release and that, if known by him or her, would have materially affected his or her settlement with the debtor or released party.” (Emphasis added.)
C.By entering into this release, Borrower recognizes that no facts or representations are ever absolutely certain and it may hereafter discover facts in addition to or different from those which it presently knows or believes to be true, but that it is the intention of Borrower hereby to fully, finally and forever settle and release all Released Claims; accordingly, if Borrower should subsequently discover that any fact that it relied upon in entering into this release was untrue, or that any understanding of the facts was incorrect, Borrower shall not be entitled to set aside this release by reason thereof, regardless of any claim of mistake of fact or law or any other circumstances whatsoever. Borrower acknowledges that it is not relying upon and has not relied upon any representation or statement made by Bank with respect to the facts underlying this release or with regard to any of such party’s rights or asserted rights.
D.This release may be pleaded as a full and complete defense and/or as a cross-complaint or counterclaim against any action, suit, or other proceeding that may be instituted, prosecuted or attempted in breach of this release. Borrower acknowledges that the release contained herein constitutes a material inducement to Bank to enter into this Amendment, and that Bank would not have done so but for Bank’s expectation that such release is valid and enforceable in all events.
E.Borrower hereby represents and warrants to Bank, and Bank is relying thereon, as follows:
1Except as expressly stated in this Amendment, neither Bank nor any agent, employee or representative of Bank has made any statement or representation to Borrower regarding any fact relied upon by Borrower in entering into this Amendment.
2Borrower has made such investigation of the facts pertaining to this Amendment and all of the matters appertaining thereto, as it deems necessary.
3The terms of this Amendment are contractual and not a mere recital.
4This Amendment has been carefully read by Borrower, the contents hereof are known and understood by Borrower, and this Amendment is signed freely, and without duress, by Borrower.
5Borrower represents and warrants that it is the sole and lawful owner of all right, title and interest in and to every Released Claim, and that it has not heretofore assigned or transferred, or purported to assign or transfer, to any person, firm or entity any Released Claim. Borrower shall indemnify Bank, defend and hold it harmless from and against all claims based upon or arising in connection with prior assignments or purported assignments or transfers of any claims or matters released herein.
9.Integration. This Amendment and the Loan Documents represent the entire agreement about this subject matter and supersede prior negotiations or agreements. All prior agreements, understandings, representations, warranties, and negotiations between the parties about the subject matter of this Amendment and the Loan Documents merge into this Amendment and the Loan Documents.
10.Counterparts. This Amendment may be executed in any number of counterparts and all of such counterparts taken together shall be deemed to constitute one and the same instrument.
11.Effectiveness. This Amendment shall be deemed effective upon (a) the due execution and delivery to Bank of this Amendment by each party hereto, and (b) Borrower’s payment to Bank of Bank’s legal fees and expenses incurred in connection with the negotiation and preparation of this Amendment.
[Signature page follows.]
IN WITNESS WHEREOF, the parties hereto have caused this Amendment to be duly executed and delivered as of the date first written above.
BANK |
| BORROWER | ||
| | | ||
SILICON VALLEY BANK | | OMEROS CORPORATION | ||
| | | ||
| | | ||
By: | /s/ Max Eberhart | | By: | /s/ Michael Jacobsen |
Name: | Max Eberhart | | Name: | Michael A. Jacobsen |
Title: | Vice President | | Title: | Chief Accounting Officer |
Schedule 1
EXHIBIT B
COMPLIANCE CERTIFICATE
TO: | SILICON VALLEY BANK | Date: | |
FROM: | OMEROS CORPORATION | | |
The undersigned authorized officer of OMEROS CORPORATION (“Borrower”) certifies that under the terms and conditions of the Loan and Security Agreement between Borrower and Bank (the “Agreement”), (1) Borrower is in complete compliance for the period ending _______________ with all required covenants except as noted below, (2) there are no Events of Default, (3) all representations and warranties in the Agreement are true and correct in all material respects on this date except as noted below; provided, however, that such materiality qualifier shall not be applicable to any representations and warranties that already are qualified or modified by materiality in the text thereof; and provided, further that those representations and warranties expressly referring to a specific date shall be true, accurate and complete in all material respects as of such date, (4) Borrower, and each of its Subsidiaries, has timely filed all required tax returns and reports, and Borrower has timely paid all foreign, federal, state and local taxes, assessments, deposits and contributions owed by Borrower except as otherwise permitted pursuant to the terms of Section 5.9 of the Agreement, and (5) no Liens have been levied or claims made against Borrower or any of its Subsidiaries, if any, relating to unpaid employee payroll or benefits of which Borrower has not previously provided written notification to Bank. Attached are the required documents supporting the certification. The undersigned certifies that these are prepared in accordance with GAAP consistently applied from one period to the next except as explained in an accompanying letter or footnotes. The undersigned acknowledges that no borrowings may be requested at any time or date of determination that Borrower is not in compliance with any of the terms of the Agreement, and that compliance is determined not just at the date this certificate is delivered. Capitalized terms used but not otherwise defined herein shall have the meanings given them in the Agreement.
Please indicate compliance status by circling Yes/No under “Complies” column.
Reporting Covenants | Required | Complies |
| | |
Quarterly financial statements | Quarterly within 45 days (Q1-Q3) | Yes No |
Compliance Certificate | When Advance outstanding, Monthly within 30 days When Advance not outstanding, Quarterly within 30 days | Yes No |
Annual financial statements (CPA Audited) | FYE within 90 days | Yes No |
10-Q, 10-K and 8-K | Within 5 days after filing with SEC | Yes No |
A/R, A/P Agings & sell-through reports | When Advance outstanding, Monthly within 30 days When Advance not outstanding, quarterly within 30 days | Yes No |
Royalty Payment Report & Borrowing Base Report | When Advance requested and at all times when Advance outstanding and streamline period is in effect, monthly within 7 days; when not in Streamline and when no Advances outstanding, quarterly within 45 days | Yes No |
Board approved projections | FYE within 90 days and as amended/updated | Yes No |
Streamline Period | Required | Actual | Eligible |
Maintain: | | | |
Liquidity | >$30,000,000.00 | $ | Yes No |
The following are the exceptions with respect to the certification above: (If no exceptions exist, state “No exceptions to note.”)
OMEROS CORPORATION |
| BANK USE ONLY | ||||
| | | ||||
| | Received by: | | |||
| | | AUTHORIZED SIGNER | |||
By: | | | | | ||
Name: | | | Date: | | ||
Title: | | | | | ||
| | | Verified: | | ||
| | | | AUTHORIZED SIGNER | ||
| | | Date: | | ||
| | | | | ||
| | | Compliance Status: | Yes | No | |
Schedule 2
Purchase Agreement
Exhibit 23.1
Consent of Independent Registered Public Accounting Firm
We consent to the incorporation by reference in the Registration Statements (Form S-8 Nos. 333-162732, 333-165861, 333-172905, 333-180216, 333-187344, 333-194693, 333-202788, 333-210219, 333-216749, 333-218882, 333-232071 and 333-257148) pertaining to the Omeros Corporation 2008 Equity Incentive Plan, the Omeros Corporation Second Amended and Restated 1998 Stock Option Plan, the nura, Inc. 2003 Stock Option Plan, the Omeros Corporation Stock Option Grant to Gregory A. Demopulos, M.D., the Omeros Corporation Stock Option Grant to Pamela Pierce Palmer, M.D., Ph.D., and the Omeros Corporation 2017 Omnibus Incentive Compensation Plan, and the Registration Statement (Form S-3 No. 333- 235349) and related Prospectus of Omeros Corporation pertaining to the registration of common stock, preferred stock, debt securities, depositary shares, warrants, subscription rights, and units, of our reports dated March 1, 2022, with respect to the consolidated financial statements of Omeros Corporation, and the effectiveness of internal control over financial reporting of Omeros Corporation, included in this Annual Report (Form 10-K) of Omeros Corporation for the year ended December 31, 2021.
/s/ Ernst & Young LLP
Seattle, Washington
March 1, 2022
Exhibit 31.1
CERTIFICATION OF PRINCIPAL EXECUTIVE OFFICER PURSUANT TO RULE 13a-14(a)/15d-14(a) OF THE SECURITIES EXCHANGE ACT OF 1934, AS ADOPTED PURSUANT TO SECTION 302 OF THE SARBANES-OXLEY ACT OF 2002
I, Gregory A. Demopulos, M.D., certify that:
1. I have reviewed this annual report on Form 10-K of Omeros Corporation;
2. Based on my knowledge, this report does not contain any untrue statement of a material fact or omit to state a material fact necessary to make the statements made, in light of the circumstances under which such statements were made, not misleading with respect to the period covered by this report.
3. Based on my knowledge, the financial statements, and other financial information included in this report, fairly present in all material respects the financial condition, results of operations and cash flows of the registrant as of, and for, the periods presented in this report;
4. The registrant’s other certifying officer(s) and I are responsible for establishing and maintaining disclosure controls and procedures (as defined in Exchange Act Rules 13a-15(e) and 15d-15(e)) and internal control over financial reporting (as defined in Exchange Act Rules 13a-15(f) and 15d-15(f)) for the registrant and have:
a. Designed such disclosure controls and procedures, or caused such disclosure controls and procedures to be designed under our supervision, to ensure that material information relating to the registrant, including its consolidated subsidiaries, is made known to us by others within those entities, particularly during the period in which this report is being prepared;
b. Designed such internal control over financial reporting, or caused such internal control over financial reporting to be designed under our supervision, to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles;
c. Evaluated the effectiveness of the registrant’s disclosure controls and procedures and presented in this report our conclusions about the effectiveness of the disclosure controls and procedures, as of the end of the period covered by this report based on such evaluation; and
d. Disclosed in this report any change in the registrant’s internal control over financial reporting that occurred during the registrant’s most recent fiscal quarter (the registrant’s fourth fiscal quarter in the case of an annual report) that has materially affected, or is reasonably likely to materially affect, the registrant’s internal control over financial reporting; and
5. The registrant’s other certifying officer(s) and I have disclosed, based on our most recent evaluation of internal control over financial reporting, to the registrant’s auditors and the audit committee of the registrant’s board of directors (or persons performing the equivalent functions):
a. All significant deficiencies and material weaknesses in the design or operation of internal control over financial reporting which are reasonably likely to adversely affect the registrant’s ability to record, process, summarize and report financial information; and
b. Any fraud, whether or not material, that involves management or other employees who have a significant role in the registrant’s internal control over financial reporting.
| Dated: March 1, 2022 |
|
|
| /s/ Gregory A. Demopulos |
| Gregory A. Demopulos, M.D. |
Exhibit 31.2
CERTIFICATION OF PRINCIPAL FINANCIAL OFFICER PURSUANT TO RULE 13a-14(a)/15d-14(a) OF THE SECURITIES EXCHANGE ACT OF 1934, AS ADOPTED PURSUANT TO SECTION 302 OF THE SARBANES-OXLEY ACT OF 2002
I, Michael A. Jacobsen, certify that:
1. I have reviewed this annual report on Form 10-K of Omeros Corporation;
2. Based on my knowledge, this report does not contain any untrue statement of a material fact or omit to state a material fact necessary to make the statements made, in light of the circumstances under which such statements were made, not misleading with respect to the period covered by this report.
3. Based on my knowledge, the financial statements, and other financial information included in this report, fairly present in all material respects the financial condition, results of operations and cash flows of the registrant as of, and for, the periods presented in this report;
4. The registrant’s other certifying officer(s) and I are responsible for establishing and maintaining disclosure controls and procedures (as defined in Exchange Act Rules 13a-15(e) and 15d-15(e)) and internal control over financial reporting (as defined in Exchange Act Rules 13a-15(f) and 15d-15(f)) for the registrant and have:
a. Designed such disclosure controls and procedures, or caused such disclosure controls and procedures to be designed under our supervision, to ensure that material information relating to the registrant, including its consolidated subsidiaries, is made known to us by others within those entities, particularly during the period in which this report is being prepared;
b. Designed such internal control over financial reporting, or caused such internal control over financial reporting to be designed under our supervision, to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles;
c. Evaluated the effectiveness of the registrant’s disclosure controls and procedures and presented in this report our conclusions about the effectiveness of the disclosure controls and procedures, as of the end of the period covered by this report based on such evaluation; and
d. Disclosed in this report any change in the registrant’s internal control over financial reporting that occurred during the registrant’s most recent fiscal quarter (the registrant’s fourth fiscal quarter in the case of an annual report) that has materially affected, or is reasonably likely to materially affect, the registrant’s internal control over financial reporting; and
5. The registrant’s other certifying officer(s) and I have disclosed, based on our most recent evaluation of internal control over financial reporting, to the registrant’s auditors and the audit committee of the registrant’s board of directors (or persons performing the equivalent functions):
a. All significant deficiencies and material weaknesses in the design or operation of internal control over financial reporting which are reasonably likely to adversely affect the registrant’s ability to record, process, summarize and report financial information; and
b. Any fraud, whether or not material, that involves management or other employees who have a significant role in the registrant’s internal control over financial reporting.
| Dated: March 1, 2022 |
|
|
| /s/ Michael A. Jacobsen |
| Michael A. Jacobsen |
Exhibit 32.1
CERTIFICATION OF PRINCIPAL EXECUTIVE OFFICER PURSUANT TO 18 U.S.C. SECTION 1350, AS ADOPTED PURSUANT TO SECTION 906 OF THE SARBANES-OXLEY ACT OF 2002
In connection with the annual report on Form 10-K of Omeros Corporation (the “Company”) for the fiscal year ended December 31, 2021, as filed with the Securities and Exchange Commission on the date hereof (the “Report”), the undersigned officer of the Company certifies, pursuant to 18 U.S.C. Section 1350, as adopted pursuant to Section 906 of the Sarbanes-Oxley Act of 2002, that:
(1) The Report fully complies with the requirements of Section 13(a) or 15(d) of the Securities Exchange Act of 1934; and
(2) The information contained in the Report fairly presents, in all material respects, the financial condition and results of operations of the Company.
This certification accompanies the Report pursuant to Section 906 of the Sarbanes-Oxley Act of 2002 and shall not, except to the extent required by the Sarbanes-Oxley Act of 2002, be deemed filed by the Company for purposes of Section 18 of the Securities Exchange Act of 1934, as amended (the “Exchange Act”), or incorporated by reference in any filing under the Securities Act of 1933, as amended, or the Exchange Act, except as may be expressly set forth by specific reference in such filing.
| Dated: March 1, 2022 |
|
|
| /s/ Gregory A. Demopulos |
| Gregory A. Demopulos, M.D. |
Exhibit 32.2
CERTIFICATION OF PRINCIPAL FINANCIAL OFFICER PURSUANT TO 18 U.S.C. SECTION 1350, AS ADOPTED PURSUANT TO SECTION 906 OF THE SARBANES-OXLEY ACT OF 2002
In connection with the annual report on Form 10-K of Omeros Corporation (the “Company”) for the fiscal year ended December 31, 2021, as filed with the Securities and Exchange Commission on the date hereof (the “Report”), the undersigned officer of the Company certifies, pursuant to 18 U.S.C. Section 1350, as adopted pursuant to Section 906 of the Sarbanes-Oxley Act of 2002, that:
(1) The Report fully complies with the requirements of Section 13(a) or 15(d) of the Securities Exchange Act of 1934; and
(2) The information contained in the Report fairly presents, in all material respects, the financial condition and results of operations of the Company.
This certification accompanies the Report pursuant to Section 906 of the Sarbanes-Oxley Act of 2002 and shall not, except to the extent required by the Sarbanes-Oxley Act of 2002, be deemed filed by the Company for purposes of Section 18 of the Securities Exchange Act of 1934, as amended (the “Exchange Act”), or incorporated by reference in any filing under the Securities Act of 1933, as amended, or the Exchange Act, except as may be expressly set forth by specific reference in such filing.
| Dated: March 1, 2022 |
|
|
| /s/ Michael A. Jacobsen |
| Michael A. Jacobsen |